

Case report
Case of Retinitis Pigmentosa a 33-year-old male presented with a chief complaint of gradual painless diminution of vision in his right eye for the last 12 years. There were no other complaints apart from vision loss.
There was no history of trauma and any other systemic problems that could be suggestive of secondary pigmentary retinopathy.
He was diagnosed with retinitis pigmentosa in the right eye 6 years ago in a tertiary eye hospital. There was no similar eye problem in his family members.
On examination, his presenting visual acuity was 4/60 in RE and 6/6 in the left eye. Visual acuity didn’t improve in RE even with pinhole or best refractive correction.
On Slit-lamp examination, the anterior segment of both eyes was normal.
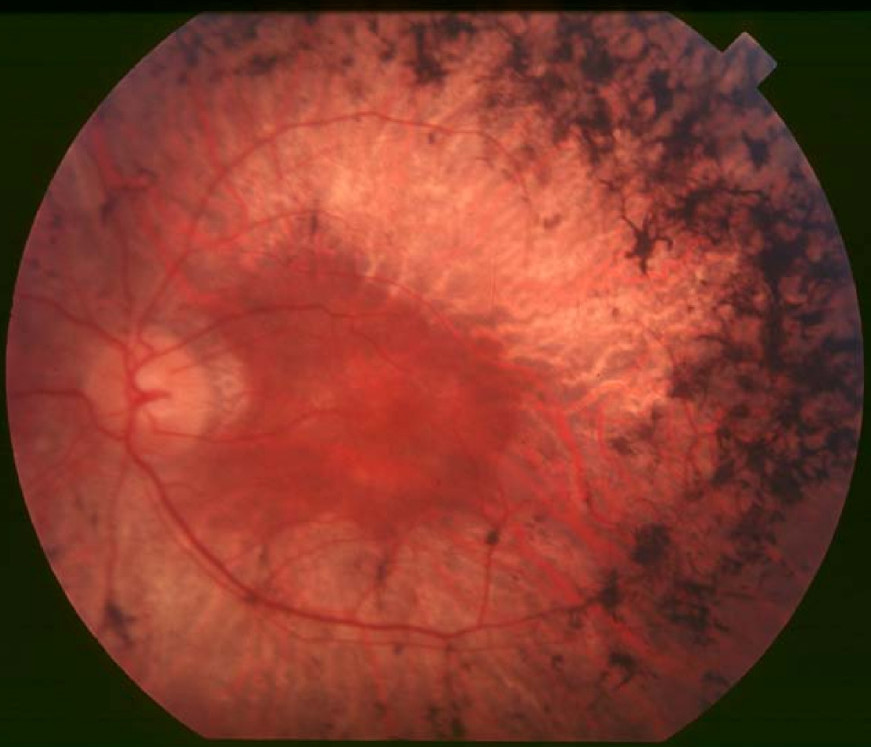
On posterior segment examination, fundus evaluation of the right eye revealed mild attenuation of blood vessels and bony spicules in the peripheral and mid-peripheral areas
whereas,
fundus examination of the left eye was normal. The intraocular pressure of both eyes was within the reference limit.
His visual field investigation showed highly decreased retinal sensitivity in the right eye whereas it was within the normal limits in the left eye.
Full-field ERG of the right eye showed decreased Rod and cone response while it was normal in the left eye.
There is a reduced amplitude for RE in all test parameters with longer latency time, suggesting functional defects in rod-cone response. With the above findings, a diagnosis of unilateral retinitis pigmentosa was made.
Disease of Retinitis Pigmentosa
Clinically and genetically is a heterogeneous group of inherited retinal disorders characterized by diffuse progressive dysfunction of predominantly rod photoreceptors with subsequent degeneration of cone photoreceptors and the retinal pigment epithelium.
Visual impairment usually manifests as night blindness and progressive visual field loss. RP may be seen in isolation (typical RP) or in association with systemic disease.
Diagnosis of Retinitis Pigmentosa
In general, the diagnosis of retinitis pigmentosa is established when the following findings are present.
- Bilateral involvement (can be asymmetric).
- Impairment of night vision and loss of peripheral vision.
- Rod dysfunction evidenced by elevated rod final threshold on dark adaptation and/or rod responses on ERG testing that are either reduced in b-wave amplitude and prolonged in implicit time or are essentially non-detectable (extinguished ERG).
- Progressive loss in photoreceptor function.
Diagnostic procedures
- Full-Field Electroretinogram (ERG)
- Dark adaptometry (DA)
- Visual field
- Electrooculogram (EOG)
- Optical coherence tomography (OCT)
- Fluorescein angiography (FA)
Management of Retinitis Pigmentosa
Many treatments have been explored without proven benefits for the isolated forms of RP. These include various vitamins and minerals, vasodilators, tissue therapy with placental extract, cortisone, cervical sympathectomy.
Also,
injections of a hydrolysate of yeast RNA, ultrasound, transfer factor, dimethyl sulfoxide, ozone, muscle transplants, and subretinal injections of fetal retinal cells.
None of the above treatments were conducted in randomized, controlled clinical trials.
It is important to note that anecdotal treatment of Retinitis Pigmentosa with a subjective improvement of the visual function should be interpreted with caution due to fluctuation in visual acuity and visual fields in this disease.
ERG is a better objective measure of remaining retinal function.
Any potential therapy will likely require several years of follow-up to assess efficacy due to the nature of the slow progression of this disease.
Read more about management strategies
would you have the interest to take Retinal imaging by your smartphone ?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera.

Read more: Morgagnian Cataract, Coats disease,Bietti Crystalline Dystrophy, Branch Retinal Artery Occlusion.
References
- Weleber RG, Gregory-Evans K. Retinitis pigmentosa and allied disorders. In: Ryan SJ, ed. Retina, 4th edn. Philadelphia, PA: Elsevier; 2006:394-485.
- Mizuno K, Nishida S: Electron microscopic studies of human retinitis pigmentosa. Part I. Two cases of advanced retinitis pigmentosa. Am J Ophthalmol. 1967;63(4):791-803.
- Bunt-Milam AH, Kalina RE, Pagon RA. Clinical-ultrastructural study of retinal dystrophy. Invest Ophthalmol Vis Sci. 1983;24(4):458-469.
- Ben-Arie-Weintrob Y, Berson EL, Dryja TP. Histopathologic-genotypic correlations in retinitis pigmentosa and allied diseases. Ophthalmic Genet. 2005;26(2):91-100.
- Pagon RA. Retinitis Pigmentosa. Surv Ophthalmol. 1988;33(3):137-177.
- Berson EL. Retinitis Pigmentosa and Allied Diseases. In: Albert D, Miller J, Azar D, Blodi B, eds. Albert and Jakobiec, 3rd edn. Philadelphia, PA: Elsevier; 2008:Ch. 177.

{kind=link}