
CASE REPORT
A 16‐year‐old girl with a retinal detachment was recruited with a known familiar history of ocular disease. The proband (III‐5) had vitreous degeneration in both eyes.
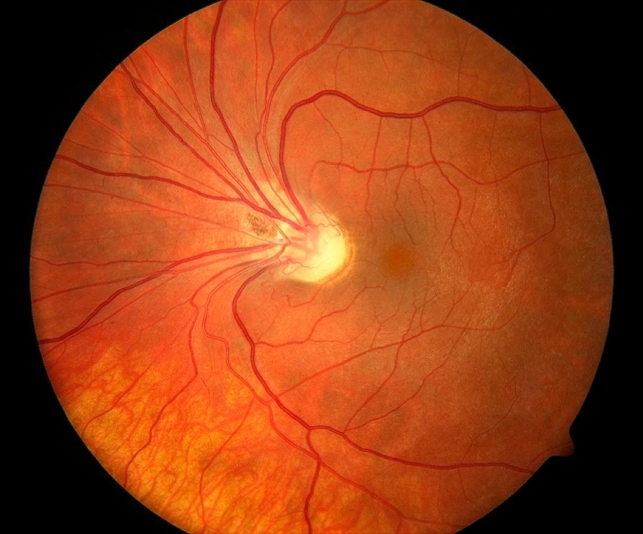
Fundus photography revealed rhegmatogenous retinal detachment in the inferior and temporal sides of her right eye, a large degeneration area in the retina, a retinal hole located at the 3 o’clock position, and proliferative cords were visible under the retina.
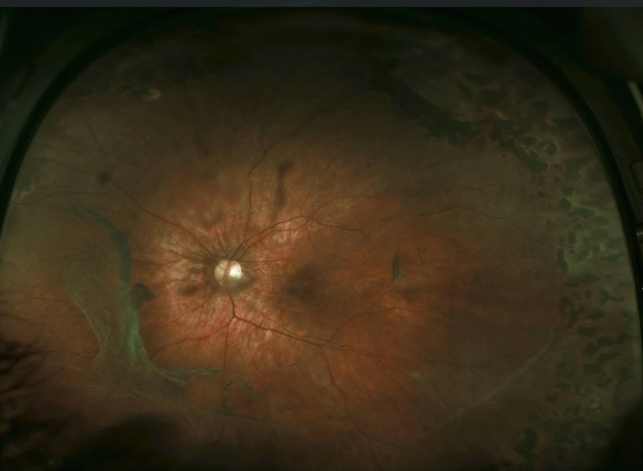
In her left eye, a large area of retinal degeneration was observed, and small blood vessels were occluded. She developed severe myopia (−6D) at the age of 15. No obvious hearing disorder was found, and there were no abnormalities in bones and joints throughout the body.
Her communication and learning capabilities were also normal. In the right eye, a vitrectomy was performed. During the operation, it was found that the vitreous and retina were closely connected without posterior vitreous detachment. After the vitreous was removed, the retina was flattened.
The hole was sealed with a laser filled with silicone oil. Six months later, silicone oil was removed and the retina remains flat. There was no significant change in the size of retinal degeneration in the left eye, in which the macular morphology was improved compared with that before the operation.
In further examination, it was found that all the patient’s family members had vitreous and retina degenerations except the patient’s mother. Glaucoma, cataracts, hypoplasia of the upper jaw, poor hearing, and joint disorders were also found in all family members.
In this family, a total of six family members over three generations had clinical diagnoses of Stickler syndrome.
DISEASE
Stickler syndrome is a systemic connective tissue disorder characterized by defective collagen production. The condition is commonly associated with ophthalmologic manifestations including vitreous abnormalities, congenital megalophthalmos, radial perivascular retinal lattice degeneration, and retinal detachment, in addition to systemic findings which may include orofacial, auditory, and musculoskeletal abnormalities.

Stickler syndrome was originally described by pediatrician Gunnar B. Stickler in 1965, as hereditary progressive arthro-ophthalmopathy.
Stickler syndrome is believed to be a direct result of abnormalities in the production of collagen types II, IX, and XI, all of which are recognized as components of the human vitreous.
Normal collagen fibrils are composed of three identical (homotrimer) or differing (heterotrimer) polypeptide chains; genetic mutations affecting the ability of consistent polypeptide chains to successfully form stable trimers, therefore, prevent the production of mature collagen and subsequently produce the clinical manifestations of Stickler syndrome.
The condition has been divided into subgroups based on ocular and systemic clinical findings, with each subgroup also corresponding to a specific genetic defect.
Subgroups of Stickler syndrome:
- Type 1 (STL1): Membranous vitreous, congenital megalophthalmos, arthropathy, deafness, Pierre Robin sequence.
- Type 2 (STL2): Fibrillar or Beaded Vitreous, otherwise similar to type 1.
- Type 3 (STL3): No ocular involvement, otherwise similar to type 1.
- Type 4 (STL4): Membranous vitreous, congenital megalophthalmos; no systemic findings.
MANAGEMENT
General treatment
Treatment of Stickler syndrome involves both prophylaxis and treatment of its complications, most commonly retinal detachment.
Medical therapy
With the possible exception of topical ocular antihypertensives for associated glaucoma, there is no effective medical treatment for prophylaxis or amelioration of the ocular signs and symptoms of Stickler syndrome.
Medical treatment by the pediatrician or primary care physician may be beneficial for the associated precocious osteoarthritis.
Surgery and Prophylaxis
Retinal detachment is a common cause of visual loss in patients with Stickler syndrome, occurring in up to 65% of affected patients. Stickler syndrome is the most common inherited cause of rhegmatogenous retinal detachment.
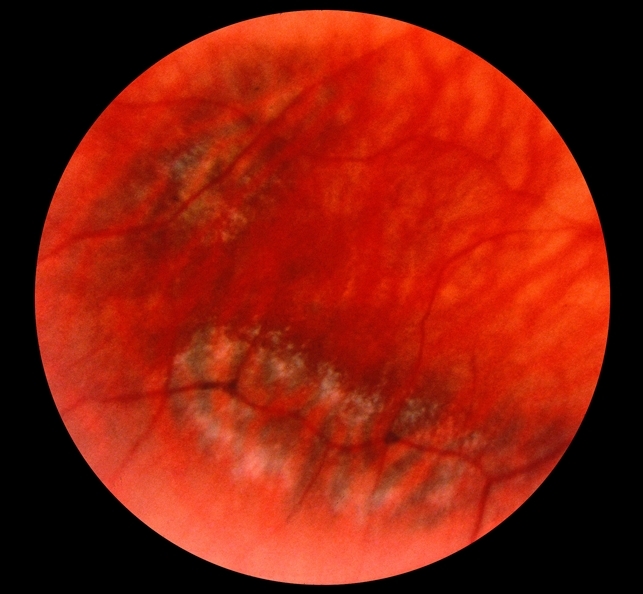
Detachment causing visual loss typically occurs in young adulthood but may occur within the first five years of life. Patients are particularly at risk of developing a giant retinal tear (GRT), which typically occurs during posterior vitreous detachment.
Associated retinal detachments are notoriously difficult to manage and patients are prone to the development of proliferative vitreoretinopathy (PVR). Treatment with primary vitrectomy is generally more successful than scleral buckling procedures and has been suggested as the procedure of choice in Stickler syndrome-related retinal detachment.
Would you have interest in taking retina images by smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
REFERENCES
- STICKLER G. HEREDITARY PROGRESSIVE ARTHRO-OPHTHALMOPATHY. Mayo Clinic proceedings. 1965-06;40:433-55.
- Snead M. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye (London). 2011-11;25:1389-400.
- Pediatric Ophthalmology and Strabismus, Section 6. Basic and Clinical Science Course, AAO, 2011-2012.
- Gurnani B, Kaur K. A rare case of stickler marshall syndrome. TNOA J Ophthalmic Sci Res 2020;58:203-5
- E.S. Parma, J. Korkko, W.S. Hagler, L. Ala-Kokko. Radial perivascular retinal degeneration: a key to the clinical diagnosis of an ocular variant of Stickler syndrome with minimal or no systemic manifestations. Am J Ophthalmol, 134 (2002), pp. 728–734
- Ziakas NG, Ramsay AS, Lynch SA, Clarke MP. Stickler’s syndrome associated with congenital glaucoma. Ophthalmic Genet. 1998 Mar;19(1):55-8. doi: 10.1076/opge.19.1.55.2177. PMID: 9587930.

{kind=link}