
CASE REPORT
A 25-year-old man was admitted for progressive visual acuity loss at night over the past 10 years. He was born into a consanguineous marriage and is the first of five siblings.
In his family, there is a positive history of deafness. In fact, the 4th male child who died at the age of 16 was complaining of nocturnal visual loss before his death; elsewhere, both parents were healthy.
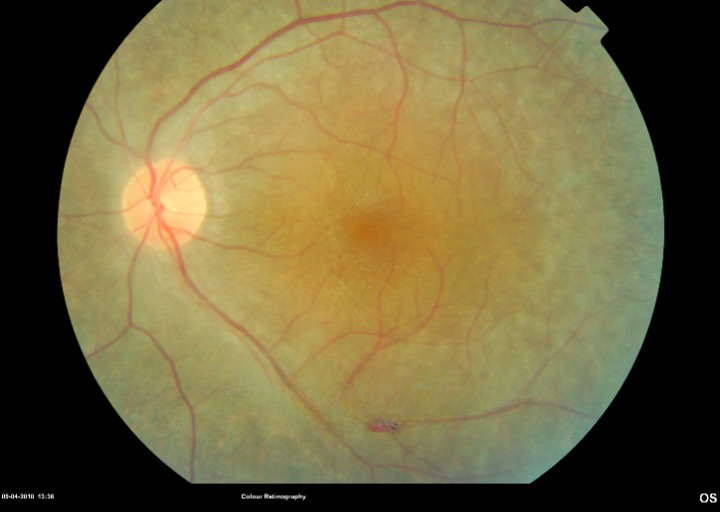
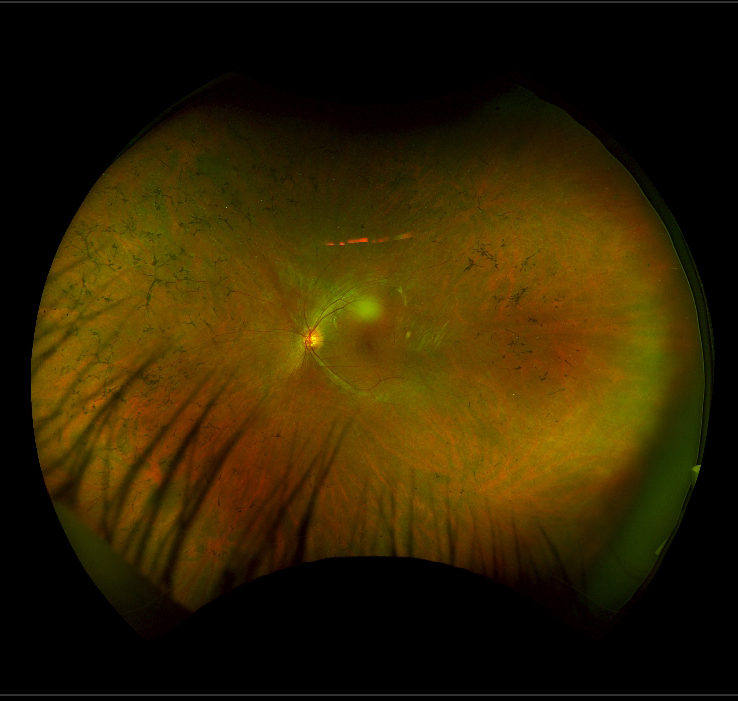
The clinical exam found a visual acuity of 6/60 in each eye. The biomicroscopic examination of the anterior segment was normal. At the fundus examination, there was a slight diffuse arteriolar narrowing, small vessels, papillary paleness, more pronounced retinal pigmented deposits on the periphery with bone spicules appearance, and macular area dystrophy.
An ERG was performed and showed a decrease in response in scotopic and photopic, confirming the degeneration of retinal visual cells (rods and cones). The neurological check-up found no abnormalities. The diagnosis of Usher syndrome type 2 was thus retained.
Usher Syndrome DISEASE entity
Usher syndrome, also known as Hallgren syndrome, is a rare genetic condition that is characterized by progressive vision and hearing loss.
Usher syndrome has been classified into three major subtypes: I, II, and III. While all three types involve progressive vision loss due to retinitis pigmentosa (RP), they are categorized according to the genes responsible and the onset and severity of the signs and symptoms.
Usher Syndrome Type I is the most severe subtype. It is characterized by profound congenital hearing loss, RP, and absent vestibular function. Usher Syndrome Type II is less severe. Patients with this subtype have moderate-to-severe congenital hearing loss, RP, and normal vestibular function.
Usher Syndrome Type III involves progressive hearing loss, RP, and varying degrees of vestibular dysfunction. Onset for type III is typically within the second to fourth decades of life. These patients also tend to have better vision than the other subtypes.
Usher Syndrome MANAGEMENT
Even though much progress has been made in elucidating the genetics and pathophysiology of Usher syndrome, there is still no cure. Treatment focuses on managing the auditory, balance, and visual manifestations.
Initial Diagnosis: The following evaluation modalities may be useful to establish the extent of the disease:
- Audiology: Otoscopy, Pure tone audiometry, assessment of speech perception
- Vestibular Function: Rotary chair, calorics, electronystagmography, and computerized posturography
- Ophthalmology: Fundoscopy, visual acuity, visual field (Goldmann perimetry), and electroretinography
- Clinical Genetics: Clinical geneticist consultation
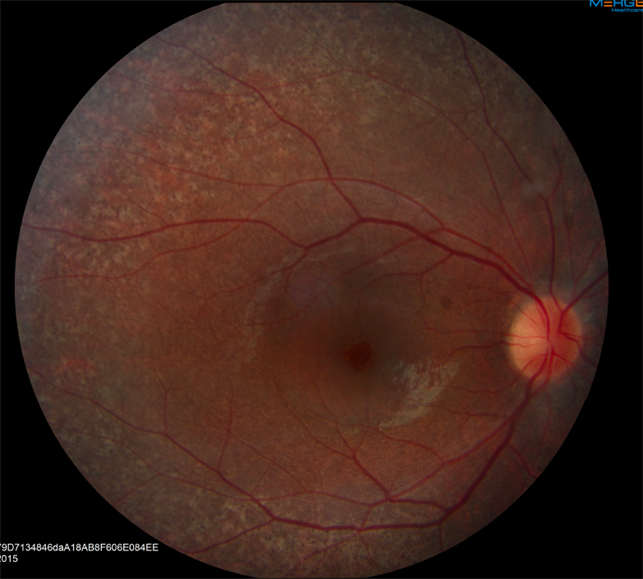
Treatment of Visual Manifestations: Visual problems result from Retinitis Pigmentosa, which is currently incurable. However, recent advances in gene therapy have led to the development of numerous trials targeting genes of interest.
Currently, the only trial actively recruiting patients is for intravitreal injection of an antisense oligonucleotide (ASO) to induce the skipping of exon 13 in USH2A. This requires patients to have a pathogenic exon 13 mutations in the USH2A gene to participate.
Besides gene therapy, management involves the use of low vision aids, portable lighting, and potentially a guide dog if visual acuity loss is severe. Various supplements such as Vitamin A, DHA (docosahexaenoic acid), and Lutein may be helpful in delaying disease progression, but the literature is equivocal on efficacy.
Surveillance: It is recommended that patients have a routine ophthalmologic evaluation to detect possible complications of the disease process such as cataracts.
Would you have interest in taking retina images by smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
REFERENCES
- Friedman TB, Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC. Usher syndrome: hearing loss with vision loss. Adv Otorhinolaryngol. 2011;70:56-65.
- Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration, and associated abnormalities. Biochim Biophys Acta. 2015;1852(3):406-20.
- Millán JM, Aller E, Jaijo T, Blanco-kelly F, Gimenez-pardo A, Ayuso C. An update on the genetics of usher syndrome. J Ophthalmol. 2011;2011:417217.
- Kimberling WJ, Hildebrand MS, Shearer AE, et al. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet Med. 2010;12(8):512-6.
- Vernon M. Usher’s syndrome–deafness and progressive blindness. Clinical cases, prevention, theory, and literature survey. J Chronic Dis. 1969;22(3):133-51.
- Boughman JA, Vernon M, Shaver KA. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis. 1983;36(8):595-603.
- Ben-yosef T, Ness SL, Madeo AC, et al. A mutation of PCDH15 among Ashkenazi Jews with the type 1 Usher syndrome. N Engl J Med. 2003;348(17):1664-70.

{kind=link}