
CASE REPORT
A 35-year-old male patient presented with a complex array of ophthalmic manifestations suggestive of Von Hippel-Lindau (VHL) Syndrome.
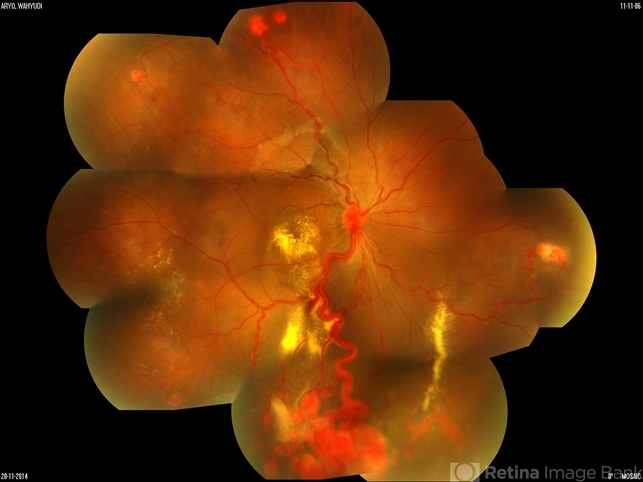
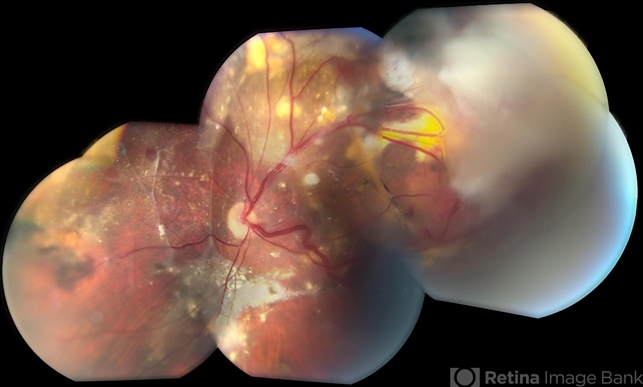
The initial clinical concern emerged when the patient reported gradual visual disturbances, prompting a meticulous ophthalmic examination.
Bilateral retinal hemangioblastomas were identified, serving as a diagnostic clue for VHL. Further imaging studies, including magnetic resonance imaging (MRI), revealed an additional cerebellar hemangioblastoma, aligning with the multisystemic nature of VHL Syndrome.
The intricate nature of this case necessitated a multidisciplinary approach, involving not only ophthalmologists but also neurologists and genetic counselors, to unravel the full spectrum of clinical implications.
Genetic testing played a pivotal role, ultimately confirming the presence of a pathogenic mutation in the VHL gene. The diagnosis of Von Hippel-Lindau Syndrome was established
DISEASE
Von Hippel-Lindau Syndrome (VHL) is a rare, autosomal dominant, familial neoplastic disease that affects the central nervous system and multiple organs such as the kidneys, pancreas, adrenals, and reproductive organs.
Mutations in the tumor suppressor gene VHL cause the disease, which commonly manifests as a variety of tumors such as hemangioblastomas of the retina and brain as well as renal cell carcinoma.
The disease affects one in every 36,000 live births and shows near-complete penetrance (90%) by 65 years of age. Cerebellar lesions, which present around age 30, are one of the most common manifestations of Von Hippel-Lindau Syndrome (VHL).
Diagnosis is confirmed with a positive familial history and at least one VHL-related tumor. However, 20% of mutations are de novo, and diagnosis for patients with a negative family history is confirmed with the occurrence of two VHL-related tumors and at least one retinal hemangioblastoma.
Retinal hemangioblastomas present the earliest around age 20 but are not present in every diagnosed case of VHL.
Von Hippel-Lindau disease manifests in the third to fourth decades of life, depending on the location of the tumors. The mean age of onset of retinal hemangioblastoma, one of the first manifestations, is around 26 years.
However, 5% of patients with retinal hemangioblastoma can present under the age of 10.
Von Hippel-Lindau disease can be split into two subtypes, Type 1 and Type 2, depending on the presence of pheochromocytomas.
Type 1 VHL has a low risk of pheochromocytomas, but both subtypes present with multiple organ tumors. Retinal hemangioblastoma usually presents bilaterally and around the optic disc.
The VHL gene is located on chromosome 3p35 and encodes the pVHL protein. One proposed mechanism of tumorigenesis for VHL involves the regulatory effect pVHL has on hypoxia-inducible factors (HIF).
Under normal conditions, von Hippel-Lindau tumor suppressor pVHL ubiquitinates HIF for degradation.
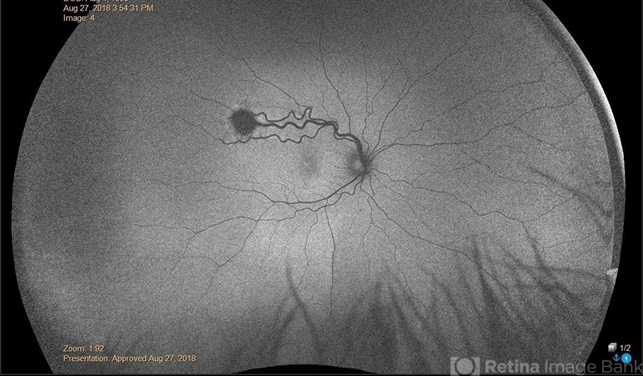
However, a lack of pVHL such as in VHL disease or hypoxia leads to stabilization of HIF-α and increased expression of tumorigenic molecules such as vascular endothelial growth factor (VEGF), platelet-derived growth factor peptide (PDGF), and transforming growth factor (TGF-a).
MANAGEMENT
No guidelines exist for the management of VHL-associated tumors, but various surgical and non-surgical interventions have been used previously.
The location, size, and degree of anatomical involvement are key elements that determine how the different neoplasms are managed.
Different lesions contain different time periods for screening, with biannual MRI scans for visceral lesions starting at age 16.
No guidelines exist for the management of VHL-associated tumors, but various surgical and non-surgical interventions have been used previously.
No guidelines exist for the management of VHL-associated tumors, but various surgical and non-surgical interventions have been used previously.
The location, size, and degree of anatomical involvement are key elements that determine how the different neoplasms are managed. Most interventions focus on shrinking or removing the tumors present in VHL disease.
CNS hemangioblastomas are standardly treated with surgical resection when they become symptomatic due to size. In instances where the neoplasm is deemed inoperable, a technique that is increasing in popularity is stereotactic radiosurgery.
New FDA-approved belzutifan, an HIF-2α inhibitor, has been used to treat neuro-associated tumors with some reports stating a 30% response in hemangioblastomas and almost half of them reducing the tumor size.
Retinal hemangioblastoma treatment is not always necessary. When it compromises vision, treatment options involve diathermy and cryocoagulation to reduce the size of retinal angiomas.
Laser photocoagulation has shown efficacy when tumors are <1.5mm. External beam radiotherapy may be used for larger tumors (diameter >4mm) which tend to be resistant to standard treatment.
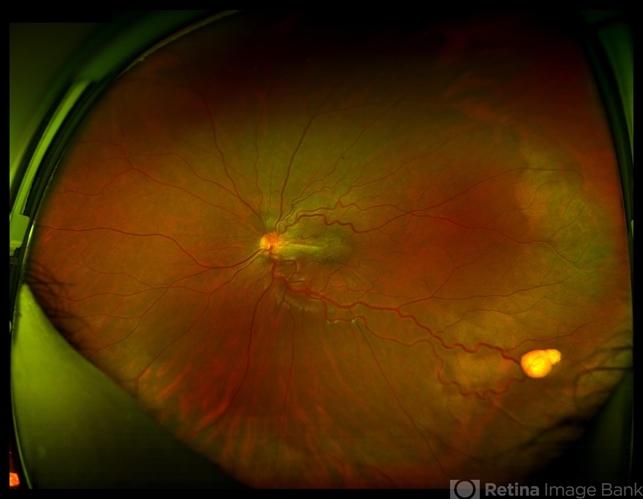
Tumors found near the optic disc may be treated with intravitreal anti-VEGF therapy if necessary.
Kidney involvement can be benign (cysts) or malignant (Renal Cell Carcinoma). Renal cell carcinomas that are 3 cm or more in diameter can be removed with a partial nephrectomy to prevent metastasis and maintain normal kidney function.
Smaller tumors could be surgically resected or undergo radiofrequency ablation or cryoablation.
Pheochromocytomas surgically managed with an adrenalectomy can prevent some of the life-threatening complications present with pheochromocytomas such as hypertension and myocardial infarction.
Endolymphatic sac tumors could be considered for resection to prevent sensorineural hearing loss and possible vestibular symptoms. Surgical resection with microsurgical techniques has proved to be an efficacious alternative.
Would you have interest in taking retinal images with your smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
REFERENCES
- Benjamin C, Yates JRW, Moore AT, et al. Clinical Features and Natural History of von Hippel-Lindau Disease. Qjm. 2012;77(2):1151-1163. doi:10.1093/qjmed/77.2.1151
- Ecol A, Desjardins RL, Latif F, et al. Identification of the von Hippel-Lindau Disease Tumor Suppressor Gene. Science (80). 1993;260:1317-1320.
- Coppin L, Plouvier P, Crépin M, Jourdain A-S, Yahya EA, Richard S, Paillerets BB-d, Cardot-Bauters C, Lejeune S, Leclerc J, Pigny P, Optimization of next-generation sequencing technologies for von Hippel Lindau (VHL) mosaic mutation detection and development of confirmation methods, The Journal of Molecular Diagnostics (2019).
- Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059-2067.
- van Leeuwaarde RS, Ahmad S, Links TP, et al. Von Hippel-Lindau Syndrome. 2000 May 17 [Updated 2018 Sep 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors.

{kind=link}