
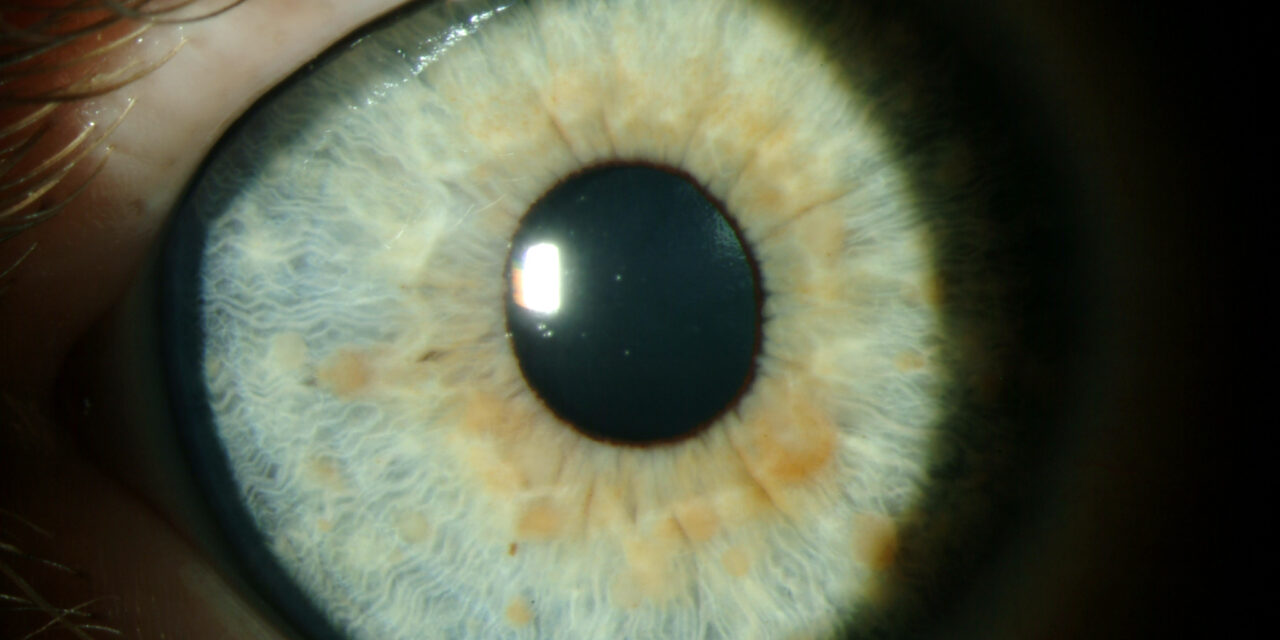
Disease Entity
Iris hamartomas are benign, focal malformations composed of disorganized but mature tissue elements native to the iris.
Rather than representing true neoplasms, these lesions arise from developmental anomalies during ocular embryogenesis.
Iris hamartomas are clinically significant not because of aggressive behavior, but because they often serve as important ocular markers of underlying systemic or genetic disorders, most notably neurocutaneous syndromes.
Although many iris hamartomas are asymptomatic and discovered incidentally, their recognition is critical, as they may prompt further systemic evaluation, genetic counseling, and long-term surveillance.
Embryology and Pathogenesis
The iris develops from both neuroectoderm (posterior pigmented epithelium and sphincter muscle) and mesenchymal neural crest cells (stroma).
Disruption of normal differentiation, migration, or maturation of these tissues during embryogenesis can result in hamartomatous growth.
Pathogenetically, iris hamartomas reflect:
-
Abnormal proliferation of native iris cells
-
Disorganized stromal architecture
-
Altered melanocyte distribution
-
Defective cellular apoptosis or signaling pathways, especially in genetic syndromes
These abnormalities are often driven by mutations affecting tumor suppressor genes or developmental signaling pathways.
Classification of Iris Hamartomas
Iris hamartomas are not a single entity but encompass several clinically distinct forms:
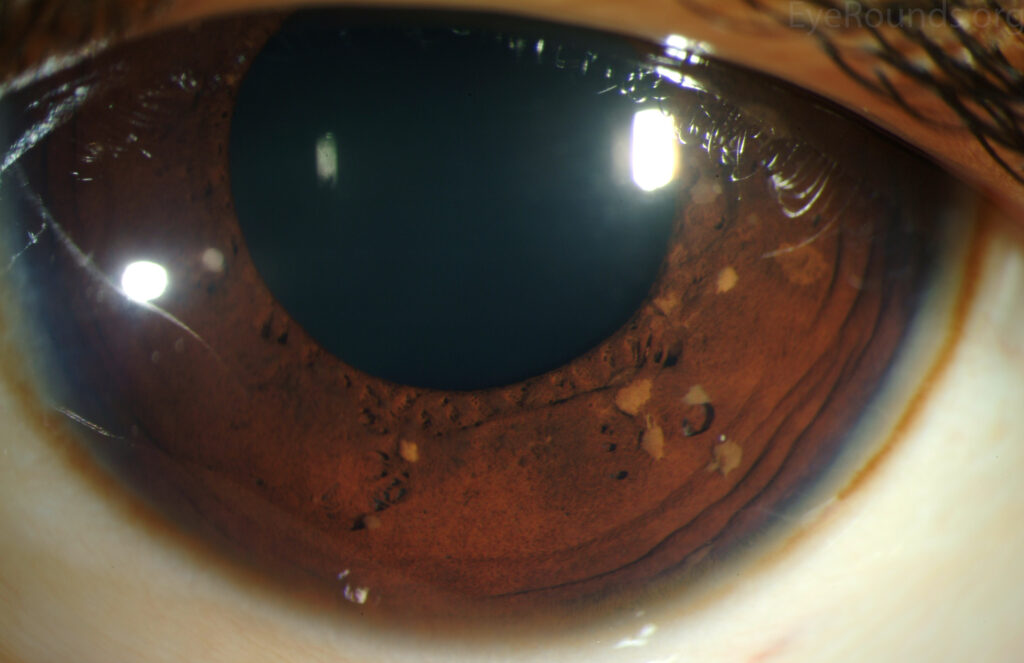
1. Lisch Nodules
Lisch nodules are the most common iris hamartomas and are considered pathognomonic for Neurofibromatosis type 1 (NF1).
-
Small, dome-shaped, well-circumscribed lesions
-
Yellow-brown or tan in color
-
Typically bilateral and multiple
-
Located on the anterior surface of the iris
-
Do not affect vision
Their number increases with age, making them useful diagnostic markers in adults with suspected NF1.
2. Iris Mammillations
Iris mammillations consist of:
-
Diffuse, evenly spaced, small conical elevations
-
Uniform pigmentation
-
Often bilateral
They are associated with:
-
Oculodermal melanocytosis
-
Congenital glaucoma
-
Increased risk of uveal melanoma
3. Iris Stromal Hamartomas
These appear as:
-
Elevated, nodular, or irregular iris lesions
-
Variable pigmentation
-
May distort iris architecture
They are less specific but may be associated with systemic hamartomatous syndromes.
4. Smooth Muscle Hamartomas
Rare lesions arising from:
-
Iris dilator or sphincter muscles
-
Typically congenital
-
May present as sectoral iris thickening or pupil distortion
Systemic Associations
The presence of iris hamartomas should prompt consideration of underlying systemic disease.
Neurofibromatosis Type 1 (NF1)
-
Autosomal dominant
-
Mutation in the NF1 gene
-
Iris hamartomas (Lisch nodules) are a diagnostic criterion
-
Associated ocular findings include optic pathway gliomas and choroidal abnormalities
Tuberous Sclerosis Complex
-
May present with iris or ciliary body hamartomas
-
Retinal astrocytic hamartomas are more common but iris involvement can occur
Oculodermal Melanocytosis
-
Associated with iris mammillations
-
Increased lifetime risk of uveal melanoma
-
Requires long-term ocular surveillance
Phakomatoses
Iris hamartomas may coexist with:
-
Sturge–Weber syndrome
-
Other neurocutaneous disorders
Clinical Presentation
Most patients are asymptomatic, and iris hamartomas are discovered during routine slit-lamp examination.
Possible clinical features include:
-
Visible iris nodules
-
Mild iris surface irregularity
-
Cosmetic iris changes
-
Rare pupil distortion or anisocoria
Visual acuity is usually unaffected unless secondary complications develop.
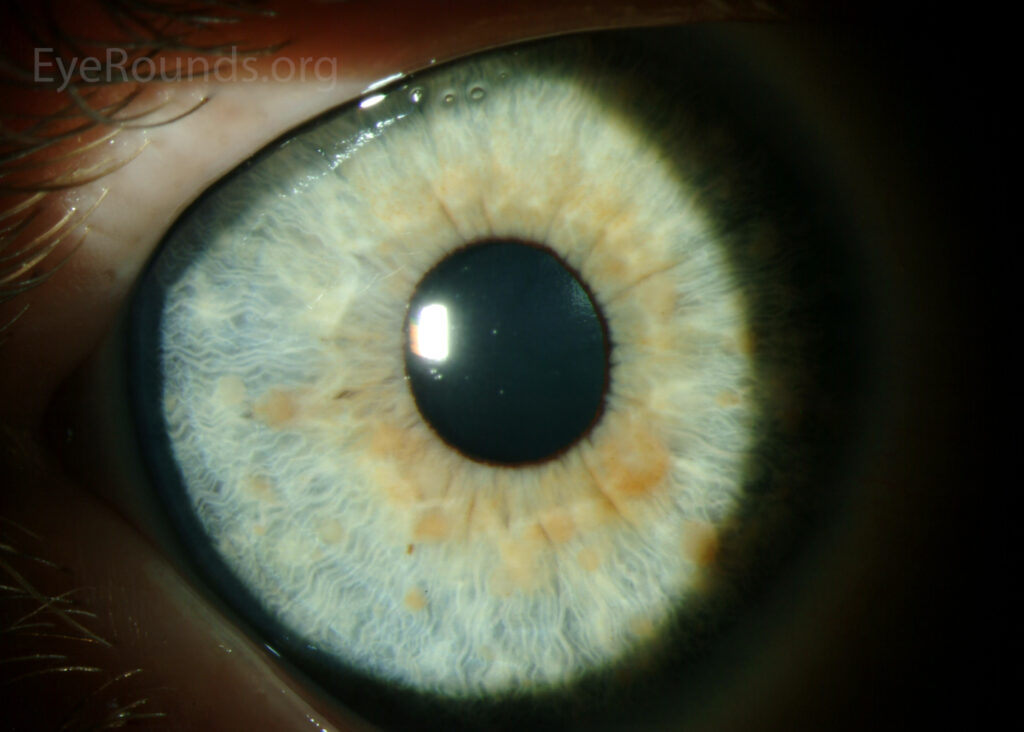
Examination Findings
Slit-Lamp Biomicroscopy
-
Well-defined nodules on the anterior iris surface
-
Smooth, dome-shaped appearance (especially Lisch nodules)
-
Absence of intrinsic vascularity
-
Stable size over time
Gonioscopy
-
Usually normal
-
Important to rule out angle involvement or secondary glaucoma
Anterior Segment Imaging
-
Anterior segment OCT can demonstrate lesion depth and stromal involvement
-
Ultrasound biomicroscopy (UBM) is useful for deeper or atypical lesions
Differential Diagnosis
Distinguishing iris hamartomas from other iris lesions is essential.
Key differentials include:
-
Iris nevus
-
Iris melanoma
-
Inflammatory iris nodules (sarcoidosis, tuberculosis)
-
Iris cysts
-
Metastatic iris tumors
Unlike neoplastic lesions, hamartomas are typically:
-
Non-progressive
-
Avascular
-
Bilateral in systemic disease
-
Stable over time
Diagnosis
Diagnosis is primarily clinical, supported by:
-
Characteristic slit-lamp findings
-
Bilaterality and multiplicity
-
Associated systemic features
Genetic testing may be indicated when:
-
Neurocutaneous syndromes are suspected
-
There is a family history
-
Systemic manifestations are present
Management
Observation
-
Most iris hamartomas require no treatment
-
Regular follow-up to ensure stability
Systemic Evaluation
-
Referral for genetic counseling if indicated
-
Dermatologic and neurologic assessment in suspected phakomatoses
Treatment of Complications
Rare complications may require intervention:
-
Secondary glaucoma
-
Iris distortion affecting pupil function
-
Diagnostic uncertainty prompting biopsy (rare)
Surgical excision is not recommended unless malignancy cannot be excluded.
Prognosis
The prognosis of iris hamartomas is excellent.
-
Lesions are benign and non-progressive
-
Visual prognosis is generally unaffected
-
Prognosis depends more on the associated systemic disease than on the iris lesion itself
Early identification improves outcomes by enabling timely diagnosis of underlying syndromes.
Importance in Ophthalmic Practice
For ophthalmologists, iris hamartomas are more than incidental findings. They serve as:
-
Diagnostic clues to systemic disease
-
Markers for genetic disorders
-
Triggers for multidisciplinary evaluation
Their recognition highlights the critical role of the eye as a window to systemic health.
Conclusion
Iris hamartomas are benign developmental anomalies of the iris that carry significant diagnostic value. While they rarely cause ocular morbidity, their association with systemic genetic disorders makes early identification essential.
A careful slit-lamp examination, awareness of characteristic features, and appropriate systemic evaluation allow ophthalmologists to play a pivotal role in the holistic care of affected patients.
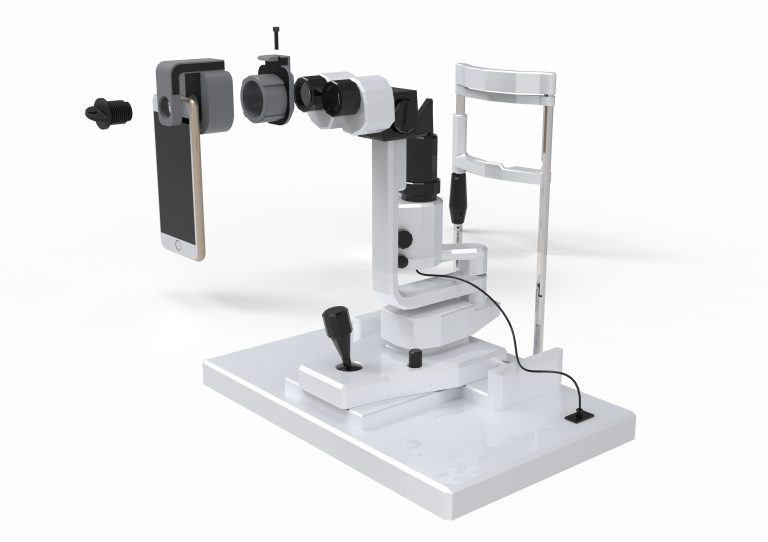
HOW TO TAKE SLIT-LAMP EXAM IMAGES WITH A SMARTPHONE?
Smartphone slit-lamp photography is a new advancement in the field of science and technology, in which photographs of the desired slit-lamp finding can be taken with smartphones by using the slit-lamp adapters.
Slit-lamp Smartphone photography
References
-
Shields JA, Shields CL. Intraocular Tumors: An Atlas and Textbook. 3rd ed. Lippincott Williams & Wilkins.
-
Eagle RC Jr. Eye Pathology: An Atlas and Text. 2nd ed. Wolters Kluwer.
-
Kanski JJ, Bowling B. Clinical Ophthalmology: A Systematic Approach. 9th ed. Elsevier.
-
Yanoff M, Duker JS. Ophthalmology. 5th ed. Elsevier.
-
Tasman W, Jaeger EA. Duane’s Ophthalmology. Lippincott Williams & Wilkins.

{kind=link}