
Case Study
A 9-year-old boy presented with progressive photophobia and decreased visual acuity in both eyes. His parents noted that he frequently squinted in bright light and complained of “sparkling” sensations in his eyes.
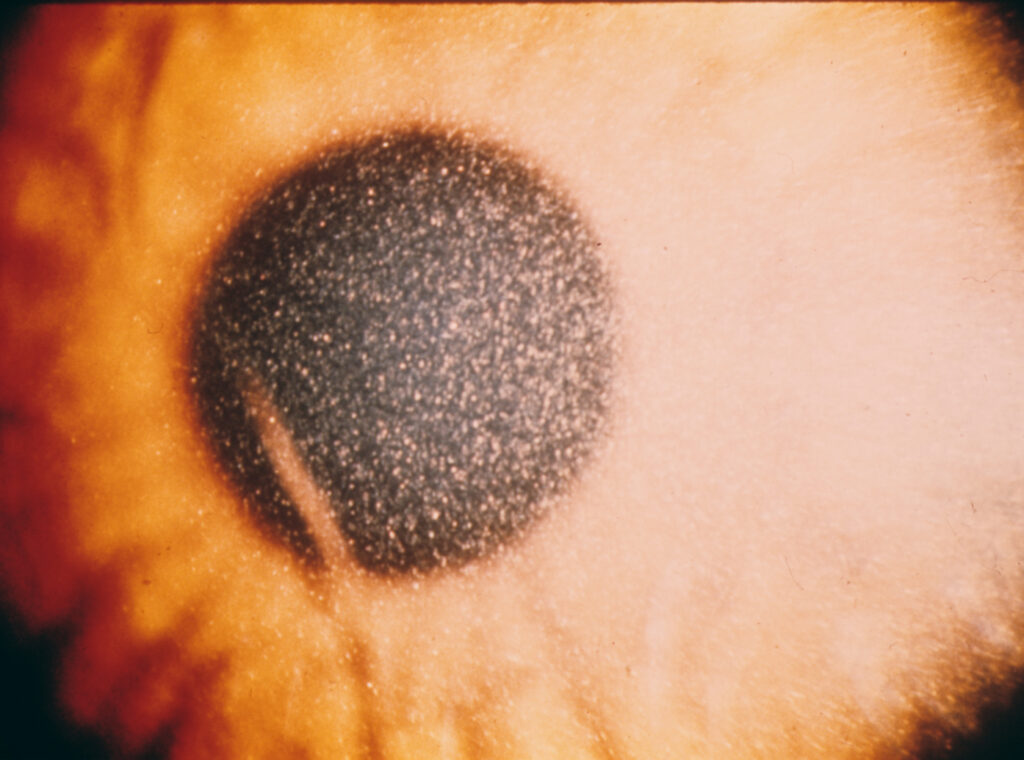
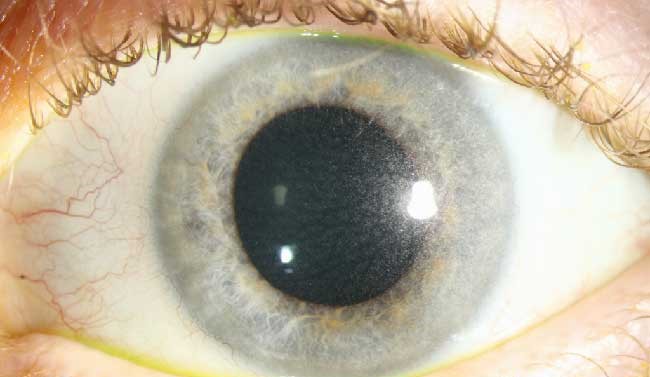
He had a history of poor growth and generalized weakness. On slit-lamp examination, multiple highly refractile, needle-shaped crystals were seen deposited throughout the corneal stroma.
Fundus examination revealed pigmentary changes in the macula.
Laboratory testing showed elevated cystine levels in leukocytes, confirming a diagnosis of Ocular Cystinosis, a rare but vision-threatening lysosomal storage disorder.
Disease Entity
Ocular Cystinosis is a manifestation of systemic cystinosis, a rare autosomal recessive lysosomal storage disease caused by mutations in the CTNS gene on chromosome 17p13.
The gene encodes cystinosin, a lysosomal membrane transporter responsible for cystine efflux.
When this transporter is defective, cystine accumulates inside lysosomes, forming crystals that deposit in various tissues—including the cornea, conjunctiva, iris, retina, and optic nerve—resulting in a range of ocular complications.
Pathophysiology
Cystinosis is fundamentally a disorder of cystine transport. In normal cells, cystine—formed from disulfide-linked cysteine molecules—is transported out of lysosomes by cystinosin.
Mutations in CTNS impair this process, leading to intralysosomal cystine accumulation and crystal formation. Over time, these crystals disrupt cellular structures, impairing normal tissue function.
In the eye:
-
Corneal deposition begins in the peripheral stroma and progresses centrally.
-
Conjunctival and iris deposits may occur in later stages.
-
Retinal pigment epithelium (RPE) and optic nerve involvement can lead to pigmentary retinopathy and optic atrophy, contributing to visual loss.
Epidemiology
Cystinosis has an estimated incidence of 1 in 100,000 to 200,000 live births. It affects both genders equally and is more common in individuals of European descent.
There are three clinical subtypes:
-
Nephropathic (Infantile) Cystinosis – the most severe and common form, presenting in infancy.
-
Juvenile (Intermediate) Cystinosis – presents in adolescence with slower progression.
-
Non-Nephropathic (Ocular) Cystinosis – limited to ocular manifestations without systemic disease.

Clinical Features
The ocular symptoms of cystinosis are typically the earliest and most persistent findings.
Common manifestations include:
-
Photophobia: due to light scattering from corneal crystals.
-
Decreased visual acuity: progressive, secondary to corneal opacity or retinal degeneration.
-
Foreign body sensation and tearing: from corneal irritation.
-
Blepharospasm: frequent in children with dense crystal deposition.
-
Retinopathy: pigmentary changes, especially in patients with nephropathic cystinosis.
-
Optic atrophy: late manifestation contributing to irreversible vision loss.
Examination Findings
Anterior Segment:
-
Slit-lamp examination reveals highly refractile, needle-like crystals in the corneal stroma, beginning peripherally and progressing centrally.
-
Crystals may also be observed in the conjunctiva, iris, and lens.
Posterior Segment:
-
Pigmentary retinopathy with bone spicule–like pigmentation.
-
Macular atrophy or cystoid changes on OCT.
-
Optic disc pallor in advanced cases.
Imaging and Laboratory Findings:
-
In vivo confocal microscopy: quantifies corneal crystal density.
-
OCT: reveals macular thinning and RPE irregularities.
-
Leukocyte cystine assay: diagnostic gold standard for systemic confirmation.
Differential Diagnosis
Conditions that may mimic ocular cystinosis include:
-
Tyrosinemia type II: corneal opacities but without crystal formation.
-
Schnyder corneal dystrophy: cholesterol and lipid deposition.
-
Bietti crystalline dystrophy: crystalline retinal deposits with RPE atrophy.
-
Cystine crystals secondary to systemic therapy (e.g., cystine supplementation): rare and localized.
Diagnosis
Diagnosis relies on a combination of clinical findings and laboratory confirmation.
Steps include:
-
Slit-lamp examination to visualize corneal crystals.
-
Leukocyte cystine concentration measurement – elevated levels confirm systemic involvement.
-
Genetic testing for CTNS mutations.
-
OCT and fundus photography for documenting retinal involvement.
Early diagnosis is critical, as systemic therapy can delay or prevent renal failure and improve visual prognosis.
Management
Management of ocular cystinosis requires both systemic and topical therapy to reduce cystine accumulation.
1. Systemic Treatment:
-
Cysteamine bitartrate (Cystagon® or Procysbi®): oral cysteamine reduces cystine levels by converting cystine to cysteine, which exits lysosomes through alternate pathways.
-
Regular monitoring of leukocyte cystine levels guides dose adjustment.
2. Ocular Treatment:
-
Cysteamine eye drops (Cystadrops® or compounded cysteamine 0.44%): dissolve corneal crystals and relieve photophobia.
-
Frequent instillation (typically 4–6 times daily) is necessary for efficacy.
-
Lubricating drops help reduce irritation and improve comfort.
-
Sunglasses or tinted lenses alleviate photophobia.
3. Supportive Management:
-
Low-vision aids for patients with macular involvement.
-
Regular follow-up with ophthalmology and nephrology to monitor systemic and ocular status.
Prognosis
With early diagnosis and continuous cysteamine therapy, patients can maintain good vision into adulthood.
However, untreated cases may develop severe photophobia, corneal scarring, and irreversible retinal degeneration leading to blindness.
In systemic forms, renal transplantation improves survival, but ocular manifestations typically persist despite systemic improvement, necessitating lifelong topical therapy.
Prevention
There are currently no preventive measures for cystinosis, but genetic counseling is recommended for families with a known history of the disease.
Prenatal diagnosis via genetic testing can identify affected fetuses early. Compliance with cysteamine therapy is crucial in preventing disease progression.
HOW TO TAKE SLIT-LAMP EXAM IMAGES WITH A SMARTPHONE?
Smartphone slit-lamp photography is the new advancement in the field of science and technology in which photographs of the desired slit-lamp finding can be taken with smartphones by using the slit-lamp adapters.
Slit-lamp Smartphone photography
References
-
Gahl WA, Kuehl EM, Iwata F, et al. Corneal crystals in nephropathic cystinosis: Natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71(1-2):100–120.
-
Tsilou ET, Rubin BI, Reed GF, et al. Ocular manifestations of nephropathic cystinosis: Report of a longitudinal study. Ophthalmology. 2002;109(11):2077–2085.
-
Nesterova G, Gahl WA. Cystinosis: The evolution of a treatable disease. Pediatr Nephrol. 2013;28(1):51–59.
-
Liang H, Labbé A, Le Mouhaër J, et al. Long-term efficacy and safety of cysteamine eye drops in ocular cystinosis: A prospective study. Ophthalmology. 2016;123(9):2033–2042.

{kind=link}