
Case Study
A 34-year-old previously healthy male presented to the retina clinic with complaints of blurred central vision and mild metamorphopsia in both eyes for one week.
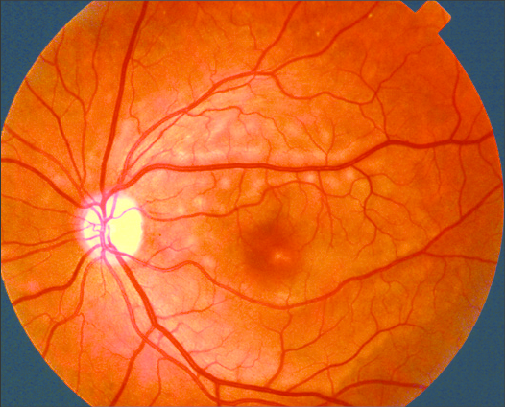
He denied ocular pain, redness, or systemic symptoms. Visual acuity was reduced to 20/40 bilaterally. Fundus examination revealed bilateral multiple round, yellowish subretinal lesions located around the macula, giving the appearance of vitelliform material.
Optical coherence tomography (OCT) revealed hyperreflective subretinal deposits and localized serous retinal detachment.
Fundus autofluorescence (FAF) showed corresponding hyperautofluorescent areas. The patient reported a recent episode of fever and malaise suggestive of a viral illness.
Based on the clinical and imaging findings, a diagnosis of Acute Exudative Polymorphous Vitelliform Maculopathy was made.
The patient was observed without intervention, and over the next three months, his vision gradually improved to 20/20, and imaging confirmed complete resolution of subretinal deposits.
Disease Entity
Acute Exudative Polymorphous Vitelliform Maculopathy (AEPVM) is a rare and self-limiting retinal disease that presents with bilateral visual symptoms and distinctive multifocal subretinal lesions resembling vitelliform deposits.
First described by Gass in the late 1980s, AEPVM affects mostly young to middle-aged adults, often following a viral prodrome.
The disorder features transient accumulation of yellow subretinal material and subretinal fluid, typically without significant intraocular inflammation or systemic illness.
Pathophysiology
The pathophysiology of AEPVM is not entirely understood. The condition is believed to arise from a temporary disruption in the function of the retinal pigment epithelium (RPE), leading to leakage of fluid and deposition of lipofuscin-like material beneath the photoreceptors.
A para-infectious autoimmune mechanism has been proposed, where an immune response following a systemic viral infection causes a reversible alteration in RPE permeability.
The serous detachment and vitelliform lesions are likely the result of accumulated photoreceptor outer segments and inflammatory byproducts.
Some theories also suggest that the condition lies within the pachychoroid spectrum due to occasional findings of choroidal thickening, but this is not universally accepted.
Epidemiology
AEPVM is exceedingly rare, with only sporadic cases reported in the literature. Both men and women are equally affected, and the age range at presentation is typically between 20 and 50 years.
Due to its acute onset and often self-resolving nature, many cases may go unrecognized or be misdiagnosed as central serous chorioretinopathy or adult-onset vitelliform dystrophy.
There is no established racial or geographic predilection, and no hereditary component has been identified.
Clinical Features
The presentation of AEPVM is relatively uniform and includes:
-
Bilateral blurry vision – Sudden or subacute in onset
-
Metamorphopsia – Reported in many cases
-
Photopsia – May occasionally occur
-
Absence of pain, redness, or photophobia
-
Normal anterior segment examination
Funduscopic examination typically shows:
-
Multiple, round, yellowish subretinal lesions around the macula
-
Shallow serous detachment
-
Absence of hemorrhages or significant vascular changes
-
No significant vitreous cells or anterior chamber inflammation
Symptoms may worsen over days to weeks but eventually stabilize and improve spontaneously.
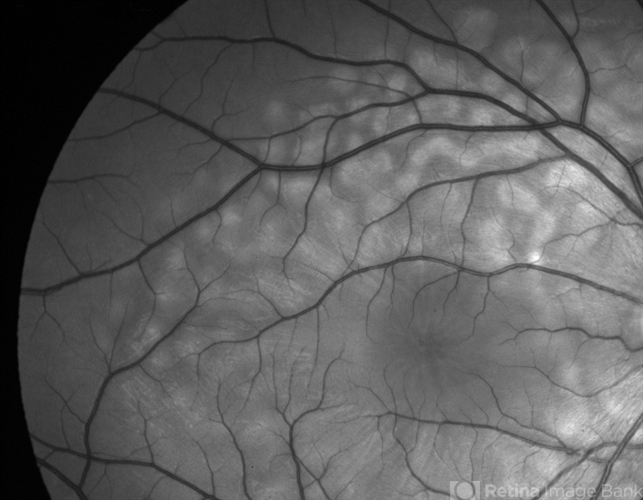
Examination Findings
Optical Coherence Tomography (OCT):
-
Hyperreflective dome-shaped subretinal deposits
-
Shallow neurosensory detachment
-
Intact external limiting membrane (ELM) and ellipsoid zone in early stages
-
Gradual resolution over weeks to months
Fundus Autofluorescence (FAF):
-
Markedly hyperautofluorescent lesions corresponding to subretinal deposits
-
Normalization with time as deposits resolve
Fluorescein Angiography (FA):
-
Mild late staining may occur
-
No significant leakage or active inflammation
Indocyanine Green Angiography (ICGA):
-
Often unremarkable or may show mild choroidal hyperpermeability
Electroretinography (ERG):
-
Typically normal, distinguishing it from hereditary dystrophies
Differential Diagnosis
Due to its appearance and symptoms, AEPVM may be mistaken for several other retinal conditions. The differential diagnosis includes:
-
Best vitelliform macular dystrophy – Usually inherited, slowly progressive, and often unilateral early on
-
Adult-onset vitelliform macular dystrophy – Similar lesion appearance but more chronic and less inflammatory
-
Vogt-Koyanagi-Harada (VKH) syndrome – Presents with bilateral serous detachments but with significant inflammation and systemic findings
-
Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE) – Involves creamy placoid lesions and often systemic malaise
-
Central Serous Chorioretinopathy (CSCR) – More focal, commonly unilateral, with ink-blot leakage on FA
-
Paraneoplastic vitelliform retinopathy – Occurs with systemic malignancies and often shows more severe progression
-
Multifocal Best disease – A hereditary condition with multifocal vitelliform lesions
Diagnosis
Diagnosis is made clinically and supported by multimodal imaging. Important features include:
-
Acute bilateral blurred vision
-
Yellowish vitelliform lesions in the macula
-
Hyperautofluorescent spots on FAF
-
Subretinal fluid and deposits on OCT
-
Recent viral prodrome in many cases
-
No signs of active intraocular inflammation
A thorough medical and ophthalmic history, combined with imaging and exclusion of hereditary or inflammatory conditions, confirms the diagnosis.
Management
There is no established treatment for AEPVM due to its self-limiting nature. Management typically involves:
-
Observation and reassurance
-
Monitoring with serial OCT and visual acuity
-
Avoiding unnecessary interventions
Some case reports have described the use of systemic corticosteroids or immunomodulatory agents in cases where inflammation is suspected or vision is significantly affected.
However, there is no conclusive evidence to support routine use.
Patients should be informed about the natural course of the disease and reassured regarding the excellent prognosis.
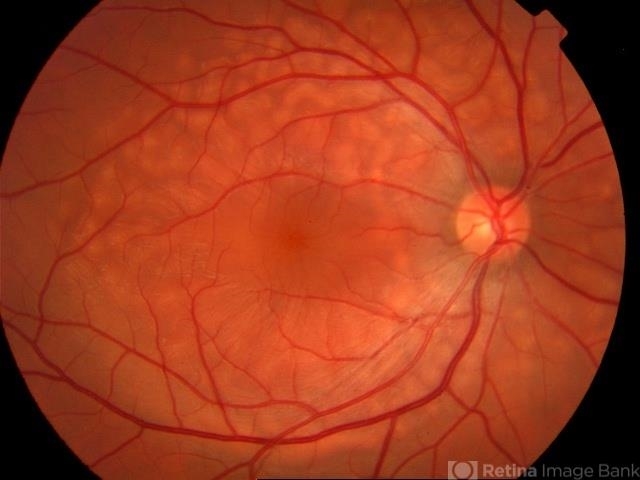
Prognosis
The long-term outlook for AEPVM is favorable. In most cases:
-
Visual acuity returns to baseline within 2–4 months
-
Subretinal deposits gradually resorb
-
No progression to chronic retinal degeneration
-
No recurrences are typically observed
Some patients may experience subtle, persistent RPE changes visible on imaging, but these rarely affect vision.
AEPVM has not been associated with systemic disease or permanent structural damage when managed conservatively.
Prevention
There are currently no preventive measures for AEPVM due to its idiopathic and acute nature.
However, increasing clinician awareness and prompt multimodal imaging help avoid unnecessary treatments and ensure accurate diagnosis.
References
-
Gass JD, et al. “Acute Exudative Polymorphous Vitelliform Maculopathy.” Archives of Ophthalmology, 1988.
-
Vaclavik V, et al. “Acute exudative polymorphous vitelliform maculopathy: Clinical features and imaging findings.” British Journal of Ophthalmology, 2011.
-
Freund KB, et al. “Multimodal Imaging in Acute Exudative Polymorphous Vitelliform Maculopathy.” Retina Today, 2017.
-
Nentwich MM, et al. “Differentiating AEPVM from Similar Conditions.” Graefe’s Archive for Clinical and Experimental Ophthalmology, 2012.

{kind=link}