
Case Study
A 21-year-old man was referred for evaluation of unusual retinal findings detected during a routine ophthalmic examination. He had no visual complaints and reported normal vision since childhood.
His past medical history was unremarkable, and there was no history of night blindness or visual field defects. Best-corrected visual acuity was 20/20 in both eyes.
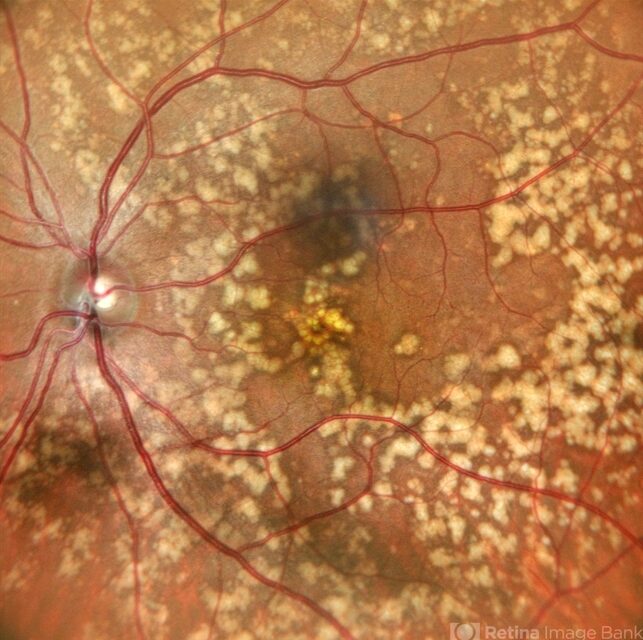
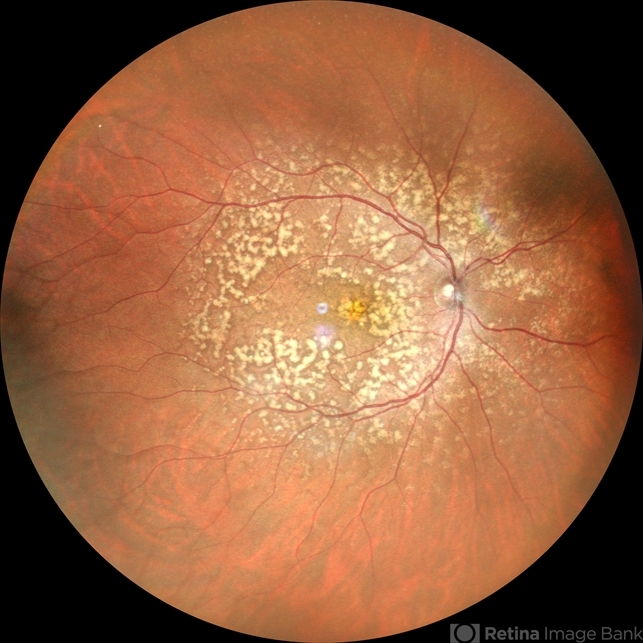
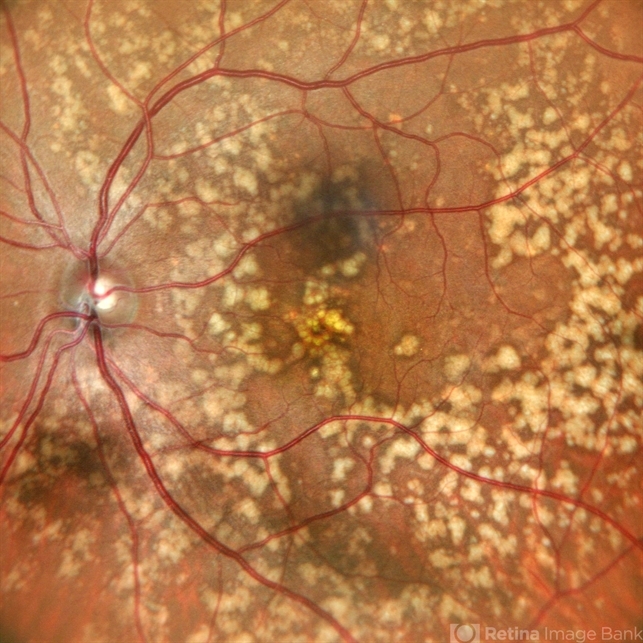
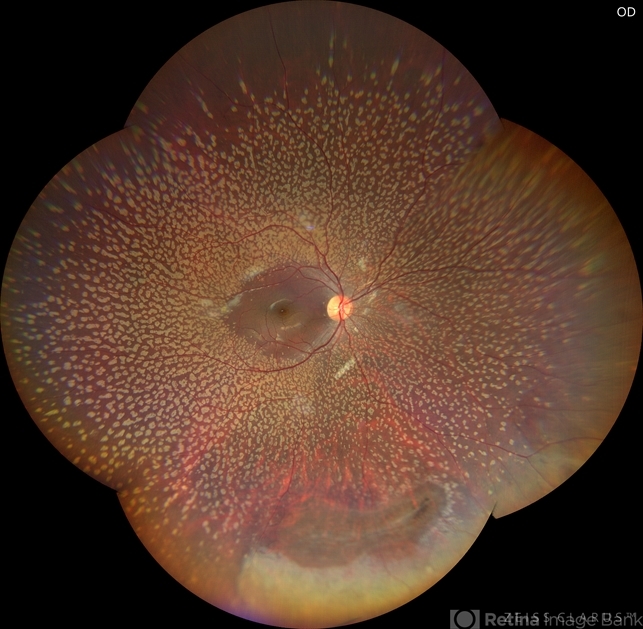
Anterior segment examination was normal. Fundus examination revealed numerous discrete, yellow-white fleck-like lesions scattered throughout the midperipheral retina in both eyes.
The lesions were located deep in the retina at the level of the retinal pigment epithelium (RPE) and spared the macula. Retinal vessels and optic discs appeared normal.
Optical coherence tomography (OCT) showed subtle hyperreflective deposits at the level of the RPE without disruption of the photoreceptor layers.
Fundus autofluorescence (FAF) demonstrated mild hyperautofluorescence corresponding to the flecks. Full-field electroretinography (ERG) and visual field testing were normal.
Further inquiry revealed that the patient’s father had similar retinal findings without visual impairment.
Based on the characteristic fundus appearance, normal retinal function, and family history, a diagnosis of benign familial fleck retina was established.
Disease Entity
Benign familial fleck retina (BFFR) is a rare inherited retinal disorder characterized by numerous yellow-white flecks distributed throughout the midperipheral retina while sparing the macula.
Despite the striking fundus appearance, visual function remains normal. The condition was first described by Krill and Klien and is considered a benign retinal phenotype with no progressive visual impairment.
BFFR belongs to a group of flecked retina disorders that also includes conditions such as fundus albipunctatus and fundus flavimaculatus.
However, unlike these disorders, BFFR does not cause night blindness or retinal dysfunction.
The lesions are typically bilateral and symmetric, affecting the retinal pigment epithelium without involvement of the central macula.
Pathophysiology
The exact pathophysiology of benign familial fleck retina is not fully understood. The disorder is believed to result from metabolic alterations within the retinal pigment epithelium, leading to the accumulation of lipofuscin-like material or other metabolic byproducts.
Histopathologic studies suggest that the flecks correspond to focal deposits at the level of the RPE or sub-RPE space.
These deposits do not appear to interfere with photoreceptor function, which explains the preservation of visual acuity and normal electrophysiologic testing.
Genetic studies have implicated mutations in the PLA2G5 gene, which encodes a secretory phospholipase involved in lipid metabolism.
Alterations in lipid processing within the RPE may lead to the accumulation of material visible clinically as flecks.
Importantly, unlike other flecked retina disorders associated with lipofuscin accumulation and retinal degeneration, BFFR does not typically cause photoreceptor damage.
Epidemiology
Benign familial fleck retina is extremely rare, with only a limited number of families reported in the literature.
The condition is inherited in an autosomal recessive pattern in many reported cases, although sporadic cases have also been described.
Both sexes appear to be affected equally. The disorder is usually detected in childhood or early adulthood during routine examination, as most patients remain asymptomatic.
Because the condition does not affect visual function, it may remain undiagnosed for many years.
Clinical Features
Most patients with benign familial fleck retina are asymptomatic.
Key clinical characteristics include:
-
Normal visual acuity
-
Absence of night blindness
-
No visual field defects
-
Bilateral symmetrical retinal flecks
The lesions are typically located in the midperipheral retina and spare the macula. They appear as:
-
Multiple yellow-white flecks
-
Round or irregular in shape
-
Deep retinal or RPE level deposits
The number and distribution of flecks may vary between individuals but generally remain stable over time.
Unlike other flecked retina disorders, BFFR does not produce progressive retinal degeneration or visual impairment.
Examination Findings
Anterior segment examination is normal.
Funduscopic findings include:
-
Numerous yellow-white flecks are scattered throughout the retina
-
Predominant involvement of the midperiphery
-
Macular sparing
-
Normal retinal vessels and optic discs
Optical Coherence Tomography (OCT)
OCT may demonstrate:
-
Small hyperreflective deposits at the level of the RPE
-
Intact ellipsoid zone and photoreceptor layers
-
Absence of retinal thinning
Fundus Autofluorescence (FAF)
-
Mild hyperautofluorescence corresponding to fleck lesions
-
Preserved macular autofluorescence
Fluorescein Angiography (FA)
-
Window defects corresponding to flecks
-
No leakage or vascular abnormalities
Electrophysiologic Testing
Full-field ERG and electro-oculography (EOG) are typically normal, confirming preserved retinal function.
Differential Diagnosis
Benign familial fleck retina must be distinguished from other conditions characterized by retinal flecks.
Important differential diagnoses include:
-
Fundus albipunctatus
-
Fundus flavimaculatus (Stargardt disease)
-
Retinitis punctata albescens
-
Pattern dystrophies of the RPE
-
Fleck retina of Kandori
Fundus albipunctatus presents with numerous white dots but is associated with congenital stationary night blindness and delayed dark adaptation.
Fundus flavimaculatus is part of the Stargardt disease spectrum and typically involves macular degeneration and progressive vision loss.
Retinitis punctata albescens is associated with progressive rod–cone dystrophy and significant ERG abnormalities.
The absence of visual symptoms and normal electrophysiologic testing are key distinguishing features of BFFR.
Diagnosis
Diagnosis of benign familial fleck retina is based on characteristic clinical findings combined with normal retinal function.
Diagnostic criteria typically include:
-
Numerous bilateral retinal flecks
-
Sparing of the macula
-
Normal visual acuity
-
Normal ERG and visual fields
-
Positive family history in some cases
Genetic testing may confirm mutations in the PLA2G5 gene, but it is not required for diagnosis.
Because the condition is benign and non-progressive, extensive systemic evaluation is usually unnecessary.
Management
No treatment is required for benign familial fleck retina.
Management consists primarily of:
-
Patient reassurance
-
Periodic ophthalmic examination
Because visual function remains normal, intervention is not necessary.
Patients should be informed about the benign nature of the condition to prevent unnecessary concern or treatment.
Genetic counseling may be considered in families with confirmed inheritance patterns.
Prognosis
The prognosis of benign familial fleck retina is excellent.
Visual acuity remains normal throughout life, and the retinal lesions typically remain stable without progression.
Unlike many other flecked retina disorders, BFFR does not lead to photoreceptor degeneration or visual impairment.
Long-term follow-up studies demonstrate preservation of retinal function and stable fundus appearance over decades.
Would you have interest in taking retinal images with your smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
References
-
American Academy of Ophthalmology. Basic and Clinical Science Course (BCSC): Retina and Vitreous. San Francisco, CA: AAO; latest edition.
-
Yanoff M, Duker JS. Ophthalmology. 5th ed. Elsevier; 2019.
-
Krill AE, Klien BA. Flecked retina syndrome. Arch Ophthalmol. 1965;74:496–508.
-
Audo I, Robson AG, Holder GE, Moore AT. Flecked retina syndromes: clinical and genetic review. Prog Retin Eye Res. 2008;27(5):540–559.
-
Traboulsi EI. Genetic Diseases of the Eye. 2nd ed. Oxford University Press; 2012.
-
Shastry BS. Benign familial fleck retina: genetic and clinical aspects. Surv Ophthalmol. 2010;55(5):503–507.

{kind=link}