
Case Study
A 14-year-old boy presented to the clinic with complaints of progressive night blindness since early childhood and increasing difficulty navigating in dim environments.
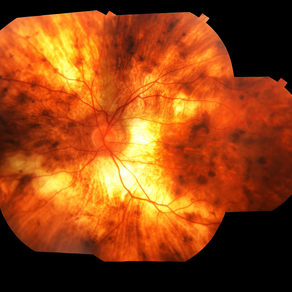
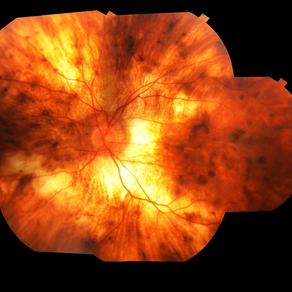
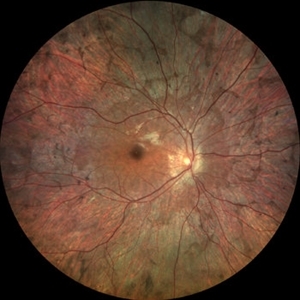
His family reported that several male relatives on his mother’s side had become legally blind in their 40s. On fundus examination, there was widespread chorioretinal atrophy with preservation of the macula.
Optical coherence tomography (OCT) showed central retinal thinning, and fundus autofluorescence imaging demonstrated a characteristic scalloped edge of residual retinal pigment epithelium (RPE).
Genetic testing confirmed a pathogenic variant in the CHM gene. The diagnosis of choroideremia was established, and the patient and family received genetic counseling and were referred for low-vision rehabilitation and ongoing clinical trial eligibility assessment.
Disease Entity
Choroideremia is a rare X-linked recessive inherited retinal dystrophy primarily affecting males and characterized by progressive degeneration of the retinal pigment epithelium (RPE), photoreceptors, and choriocapillaris.
Female carriers may show variable expression due to X-chromosome inactivation but usually have milder symptoms. The condition leads to gradual vision loss, beginning with nyctalopia (night blindness) in childhood, followed by peripheral vision loss, and eventually central vision deterioration in later decades.
Pathophysiology
Choroideremia results from mutations in the CHM gene located on Xq21.2, which encodes Rab escort protein-1 (REP1). This protein is essential for intracellular trafficking by regulating Rab GTPases involved in vesicle movement.
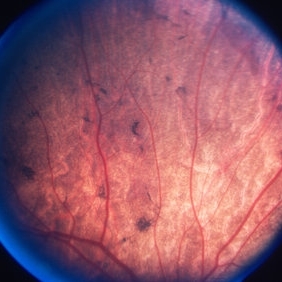
In the absence of functional REP1, cells of the RPE, photoreceptors, and choroid undergo progressive degeneration. This tri-layer loss is distinctive, with degeneration beginning in the mid-peripheral retina and moving centrally over time, often leaving an island of preserved macula until late stages.
Epidemiology
-
Prevalence: Estimated at 1 in 50,000 to 1 in 100,000 males.
-
Inheritance: X-linked recessive.
-
Gender distribution: Affects predominantly males; female carriers usually asymptomatic or mildly symptomatic.
-
Onset: Symptoms typically start in childhood or adolescence.
Clinical Features
Symptoms
-
Night blindness (nyctalopia): Often the first symptom, appearing in childhood.
-
Peripheral vision loss: Progressive over years, leading to tunnel vision.
-
Central vision: Usually preserved until the 5th or 6th decade.
-
Color vision defects: Can occur in late-stage disease.
-
Photophobia: May be present in some cases.
Signs
-
Fundus appearance:
-
Scalloped patches of chorioretinal atrophy starting in the mid-periphery
-
Sparing of the macula in early to mid-stages
-
Visibility of bare sclera in advanced disease
-
-
Optical coherence tomography (OCT): Shows progressive thinning of the outer retina and RPE
-
Fundus autofluorescence: Demonstrates areas of RPE loss with a granular peripheral autofluorescence pattern
-
Electroretinography (ERG): Shows reduced or extinguished rod and cone responses.

Examination Findings
-
Visual acuity: Normal in early stages; decreases with central involvement
-
Visual field testing: Mid-peripheral scotomas progressing to concentric narrowing
-
OCT: Retinal thinning and ellipsoid zone loss with foveal sparing
-
Autofluorescence imaging: Helps track the progression of RPE loss
-
ERG: Abnormal or absent responses, particularly rods
Differential Diagnosis
-
Retinitis pigmentosa (RP): More pigmentary changes (bone spicules), inheritance patterns vary
-
Gyrate atrophy: Autosomal recessive, ornithine metabolism disorder, elevated plasma ornithine
-
Bietti crystalline dystrophy: Crystals in the retina and cornea
-
Cone-rod dystrophies
-
Chronic uveitis with chorioretinal atrophy
Diagnosis
The diagnosis is primarily clinical, supported by multimodal imaging and confirmed by genetic testing.
Diagnostic tools:
-
Genetic testing: Detection of mutations in the CHM gene
-
OCT: Assessment of retinal layers
-
Fundus autofluorescence: Mapping of RPE integrity
-
ERG: Functional assessment of photoreceptors
-
Visual field testing: Evaluation of peripheral vision loss
Management
There is no cure yet, but ongoing research, including gene therapy trials, is showing promising results.
Current Management Strategies
-
Low vision aids: For improving the quality of life and daily functioning
-
Orientation and mobility training
-
Genetic counseling: Essential for affected individuals and family planning
-
Regular monitoring: To document disease progression and trial eligibility
-
Protection from phototoxicity: Use of sunglasses and light filters
Emerging Therapies
-
Gene therapy: Clinical trials using viral vectors (AAV2-REP1) have shown slowed degeneration and visual improvement in some patients
-
Stem cell therapy: Under investigation
-
Retinal prosthetics: In research for late-stage blindness
Prognosis
-
Slowly progressive: Most patients retain useful central vision into middle age
-
Legally blind: Many affected individuals become legally blind in their 40s to 60s
-
Variable severity: Even among family members with the same mutation
-
Carrier females: Often asymptomatic but may develop mild visual changes later in life.
Prevention
-
Currently, there is no prevention for disease onset
-
Genetic counseling and carrier detection: Important for families with known mutations
-
Prenatal and preimplantation genetic diagnosis: Options for families considering reproduction
Conclusion
Choroideremia is a rare but increasingly understood inherited retinal dystrophy. Characterized by night blindness and peripheral vision loss in males, it progresses to central vision loss in adulthood.
Advances in imaging and genetic testing allow for earlier and more accurate diagnosis, while emerging therapies—especially gene therapy—offer hope for modifying disease progression.
Early diagnosis, multidisciplinary care, and participation in clinical trials are key for optimal management and future outcomes.
Would you have interest in taking retinal images with your smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
References
-
MacDonald IM, Russell L, Chan CC. Choroideremia: New Findings from Ocular Pathology and Review of Recent Literature. Surv Ophthalmol. 2009.
-
Xue K, MacLaren RE. Gene therapy for choroideremia using an adeno-associated viral (AAV) vector. Cold Spring Harb Perspect Med. 2014.
-
van den Hurk JA, et al. The clinical spectrum of choroideremia and carrier state in the Netherlands. Br J Ophthalmol. 2007.
-
Edwards TL, et al. Assessment of Visual Function in Clinical Trials for Choroideremia. Eye. 2016.
-
Dimopoulos IS, et al. Macular Structure and Function in Choroideremia: Correlation with Disease Stage and Implications for Gene Therapy. Invest Ophthalmol Vis Sci. 2017.

{kind=link}