
CASE REPORT
A 32-year-old female presented with an acute loss of vision in her left eye, accompanied by pain exacerbated upon eye movement.

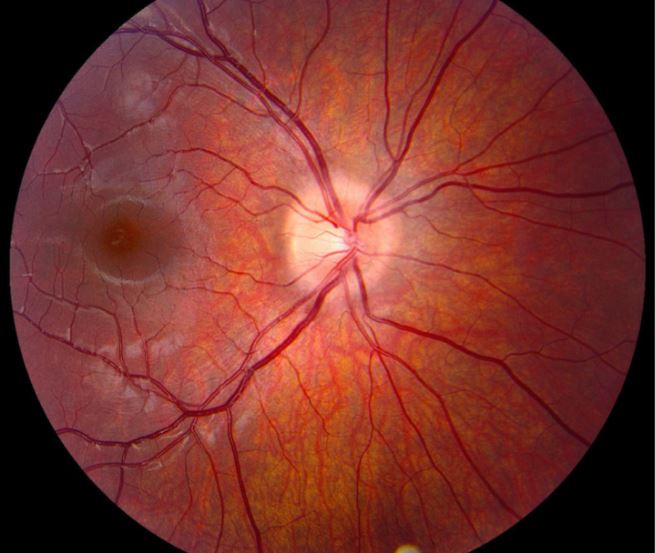
Clinical examination revealed decreased visual acuity (20/200) in the left eye, a relative afferent pupillary defect, and optic disc edema indicative of optic neuritis.
The diagnostic workup included brain and orbital magnetic resonance imaging (MRI) showing enhancement of the left optic nerve, consistent with optic neuritis.
Lumbar puncture revealed mild lymphocytic pleocytosis, elevated protein levels, and the presence of oligoclonal bands in cerebrospinal fluid.
Further MOG-IgG testing confirmed the presence of MOG-IgG antibodies, supporting the diagnosis of MOG-IgG-associated optic neuritis.
DISEASE
Myelin Oligodendrocyte Glycoprotein (MOG) Optic Neuritis is an antibody-mediated demyelinating disease of the central nervous system (CNS) that is a distinct entity from other demyelinating processes of the CNS such as Multiple Sclerosis (MS) or AQP4-Ab-associated neuromyelitis optica spectrum disorder (NMOSD).
Typical optic neuritis (ON) presents with acute, unilateral, onset of variable visual acuity/visual field loss, retrobulbar pain (worse with eye movement), loss of color vision, a relative afferent pupillary defect (RAPD), and a normal fundus exam (retrobulbar optic neuritis).
Optic neuritis in MOG however is often “atypical” and may be markedly steroid responsive, bilateral rather than unilateral, and may be associated with optic disc edema rather than retrobulbar optic neuritis.

Although optic neuritis is the most common symptom in MOG-Ab seropositive disease it can present with acute disseminated encephalomyelitis, myelitis, or an NMOSD-like presentation.
Children less than 9 years old who are positive for MOG-Antibodies (Ab) more frequently present with acute disseminated encephalomyelitis (ADEM) that may be relapsing or recurrent and may present with optic neuritis later in life.
MOG-related optic neuritis may be associated with certain HLA haplotypes. There is wide variability in the epidemiology, severity of acute vision loss, extraocular neurologic deficits, and prognosis seen in various different studies of MOG however.
Clinical diagnosis
Definitive diagnosis of MOG optic neuritis is made in the appropriate clinical setting by seropositivity of MOG-Ab (cell-based assays are the current gold standard).
One prospective cohort study of patients with optic neuritis showed that by testing all cases of optic neuritis with bilateral optic neuritis, recurrent optic neuritis, or optic disc swelling on fundoscopy for MOG antibody, all cases of MOG ON would be detected and only 50% of ON cases in the cohort would be tested overall.
An absence of each of the atypical optic neuritis features that were considered to be higher risk for MOG had a negative predictive value of over 90%.
MRI findings (e.g., OPN, lack of demyelinating white matter lesions for MS, longitudinally extensive enhancement) are also highly suggestive of MOG optic neuritis.
Imaging
Longitudinally, extensive (involving >50% of the length of the optic nerve) enhancement and/or T2-weighted hyperintensities in the anterior visual pathways can be seen in MOG-Ab optic neuritis.
Enhancing lesions are usually seen in the orbital and intracranial regions of the optic nerve. Optic perineuritis (OPN) with enhancement of the optic nerve sheath is a finding that is common in MOG optic neuritis as compared to MS-related ON (parenchymal optic nerve enhancement).
Spinal MRI findings include swelling of the spinal cord and contrast enhancement that can resemble the transverse myelitis of NMOSD.
In contrast to the ovoid, periventricular, white matter lesions (including corpus callosum) seen in MS, non-specific white matter lesions or a normal brain MRI may be seen in MOG

MANAGEMENT
Treatment
The widely accepted treatment for acute attacks has been intravenous, high-dose corticosteroid therapy (e.g., methylprednisolone) followed by intravenous immunoglobulin (IVIG) or plasma exchange in patients who do not respond to IV steroid treatment.
Methylprednisolone has been shown to increase recovery by 10-20% compared to no treatment. Of the patients who were not responsive to methylprednisolone, 40% showed improvement with IVIG, although partial recovery was more common than full recovery with IVIG.
While treatment efficacy may be low in acute attacks, there has been substantial success in preventing recurrent attacks.
In one study, 95% of patients receiving doses of at least 20 mg of prednisone for 6 months following a MOG optic neuritis attack had no recurrent episodes of optic neuritis at follow-up of over a year.
High dose and longer length of treatment was strongly associated with remittance, and patience who were given a tapered dose, or discontinued therapy earlier had relapse rates comparable to those with no treatment.
Randomized, controlled, treatment trials are limited for MOG optic neuritis, but observational open-label work suggests a role for high-dose steroids and plasma exchange in the treatment of acute attacks, and immunosuppressive therapy (e.g., steroids, oral immunosuppressants, and rituximab) as maintenance treatment.
Prognosis
In one study, 25% of patients suffering an acute attack had a permanent visual disability. In other studies, patients experienced complete or partial recovery in up to 90% of cases.

Although MOG optic neuritis attacks have been show to permanently damage the neural retinal ganglion and peripheral nerve fiber layers of the retina, the majority of patients fully or partially regain vision.
The majority of individuals in all studies have a recurrence within the first year of attack. The increased risk of permanent neurologic deficit with subsequent attacks is much worse than after the first attacks.
Younger age at first attack is associated with increased risk of permanent vision loss. Persistent MOG-Ab titers have been associated with relapse compared with patients without persistent MOG-Ab.
Would you have interest in taking retinal images with your smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
REFERENCES
- Lee, AG. Myelin Oligodendrocyte Glycoprotein (MOG). Neuro-ophthalmology Virtual Education Library: NOVEL. Creative Commons Attribution-NonCommercial-NoDerivs 2.0 Generic (CC BY-NC-ND 2.0).
- Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15(2):89-102. doi:10.1038/s41582-018-0112-x.
- In cooperation with the Neuromyelitis Optica Study Group (NEMOS), Jarius S, Ruprecht K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0
- Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin Oligodendrocyte Glycoprotein Antibody–Positive Optic Neuritis: Clinical Characteristics, Radiologic Clues, and Outcome. Am J Ophthalmol. 2018;195:8-15. doi:10.1016/j.ajo.2018.07.020.
- Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140(12):3128-3138. doi:10.1093/brain/awx276.
RETINAL IMAGING BY YOUR SMARTPHONE

RETINAL IMAGING BY YOUR SMARTPHONE
{kind=link}