
Case Study
A 38-year-old woman presented for evaluation of gradually progressive difficulty with central vision, particularly when reading in dim lighting.
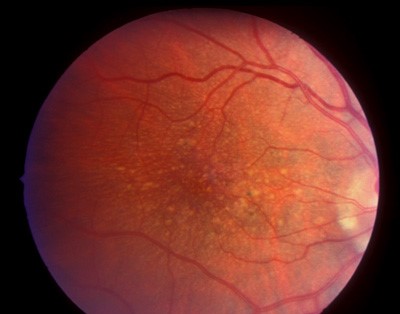
She reported that several family members, including her mother and maternal grandfather, had been diagnosed with “early macular degeneration” at relatively young ages. Best-corrected visual acuity was 20/30 in both eyes.
Anterior segment examination was normal. Fundus examination revealed numerous round, yellow-white deposits scattered throughout the posterior pole and extending into the midperipheral retina.
The drusen were relatively large and radially distributed around the macula. The optic discs and retinal vessels appeared normal.
Optical coherence tomography (OCT) demonstrated multiple dome-shaped elevations of the retinal pigment epithelium (RPE) corresponding to drusen deposits.
Fundus autofluorescence (FAF) revealed patchy hyperautofluorescence overlying the lesions. Fluorescein angiography (FA) showed early hyperfluorescence consistent with window defects without significant leakage.
Based on the characteristic drusen distribution, early onset, and positive family history, a diagnosis of autosomal dominant drusen (Malattia Leventinese) was made.
Disease Entity
Autosomal dominant drusen (ADD), also known as Malattia Leventinese or Doyne honeycomb retinal dystrophy, is a hereditary macular dystrophy characterized by the development of numerous drusen deposits in the posterior pole and midperipheral retina.
The condition is caused by mutations in the EFEMP1 gene, which encodes fibulin-3, an extracellular matrix protein involved in the structural support of Bruch’s membrane and the retinal pigment epithelium.
ADD shares several clinical features with age-related macular degeneration (AMD), particularly the presence of drusen and the risk of choroidal neovascularization.
However, it differs in its earlier age of onset, characteristic radial distribution of drusen, and clear genetic inheritance pattern.
Pathophysiology
The pathogenesis of autosomal dominant drusen involves structural abnormalities in Bruch’s membrane and the extracellular matrix between the retinal pigment epithelium and the choriocapillaris.
Mutations in the EFEMP1 gene lead to abnormal folding and accumulation of fibulin-3 protein. This results in deposition of extracellular material between the RPE and Bruch’s membrane, forming drusen-like deposits.
These deposits disrupt normal metabolic exchange between the choroid and photoreceptors, leading to progressive dysfunction of the RPE and outer retina.
Histopathologic studies demonstrate thickening of Bruch’s membrane and accumulation of hyaline material beneath the RPE. Over time, chronic metabolic stress may lead to:
-
RPE atrophy
-
Photoreceptor degeneration
-
Development of choroidal neovascularization
Although the disease shares pathologic similarities with AMD, its genetic origin and earlier onset distinguish it as a separate entity.
Epidemiology
Autosomal dominant drusen is a rare inherited retinal disorder. The disease follows an autosomal dominant inheritance pattern with high penetrance.
Affected individuals typically develop visible drusen in adolescence or early adulthood, although visual symptoms may not appear until the third or fourth decade of life.
Both sexes are affected equally. The condition was first described in families from the Leventine valley of Switzerland, giving rise to the term Malattia Leventinese. However, cases have since been reported worldwide.
Clinical Features
The clinical presentation of autosomal dominant drusen varies depending on disease stage.
Early stages are often asymptomatic, with drusen detected during routine examination.
As the disease progresses, patients may experience:
-
Mild reduction in central visual acuity
-
Metamorphopsia
-
Difficulty reading
-
Delayed dark adaptation
Characteristic fundus findings include numerous drusen arranged in a radial or honeycomb pattern around the macula.
Drusen may also extend into the midperipheral retina, which helps distinguish the disease from age-related macular degeneration.
Examination Findings
Anterior segment examination is typically normal.
Fundus examination reveals:
-
Numerous round yellow-white drusen
-
Radial distribution centered on the macula
-
Honeycomb-like pattern in advanced cases
-
Possible RPE pigmentary changes
In later stages, complications such as geographic atrophy or choroidal neovascularization may occur.
Optical Coherence Tomography (OCT)
OCT demonstrates:
-
Dome-shaped elevations of the RPE corresponding to drusen
-
Thickening of Bruch’s membrane
-
Possible outer retinal thinning in advanced stages
Fundus Autofluorescence (FAF)
FAF findings include:
-
Areas of hyperautofluorescence corresponding to drusen
-
Hypoautofluorescent areas in regions of RPE atrophy
Fluorescein Angiography (FA)
Typical findings include:
-
Early hyperfluorescence due to window defects
-
Late staining of drusen
-
Leakage if choroidal neovascularization develops
Optical Coherence Tomography Angiography (OCTA)
OCTA may detect early choroidal neovascular membranes before clinical symptoms develop.
Differential Diagnosis
Autosomal dominant drusen should be distinguished from other conditions associated with drusen deposits.
Important differential diagnoses include:
-
Age-related macular degeneration (AMD)
-
Basal laminar drusen
-
Pattern dystrophy
-
Stargardt disease
-
Cuticular drusen
Age-related macular degeneration typically occurs later in life and lacks the strong familial inheritance pattern.
Basal laminar drusen are smaller and more numerous, often producing a “starry sky” appearance on fluorescein angiography.
Pattern dystrophies may present with pigmentary changes, but do not show the characteristic radial drusen distribution seen in ADD. Family history and genetic testing can help confirm the diagnosis.
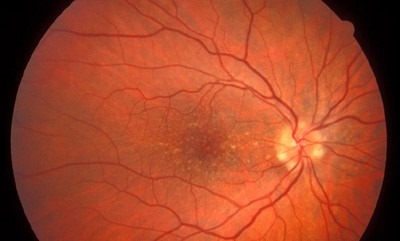
Diagnosis
Diagnosis is based on clinical examination and imaging findings.
Key diagnostic features include:
-
Early onset of drusen formation
-
Radial or honeycomb pattern of deposits
-
Bilateral involvement
-
Positive family history
Genetic testing identifying EFEMP1 mutations can confirm the diagnosis.
Multimodal imaging, including OCT and FAF, is useful for monitoring disease progression and detecting complications.
Management
There is no definitive cure for autosomal dominant drusen.
Management focuses on monitoring for complications and preserving visual function.
Recommended management strategies include:
-
Regular ophthalmic examinations
-
OCT monitoring for early detection of neovascularization
-
Patient education about symptoms of macular complications
If choroidal neovascularization develops, treatment with intravitreal anti-VEGF therapy is indicated.
Low-vision rehabilitation may be helpful in advanced stages with significant visual impairment.
Genetic counseling is recommended for affected families.
Prognosis
The prognosis of autosomal dominant drusen varies.
Many patients maintain relatively good visual acuity during early adulthood. However, progressive RPE dysfunction may eventually lead to:
-
Geographic atrophy
-
Choroidal neovascularization
-
Central vision loss
The rate of progression varies between individuals, even within the same family.
Regular follow-up is therefore essential for early detection of vision-threatening complications.
Would you have interest in taking retinal images with your smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
RETINAL IMAGING BY YOUR SMARTPHONE
References
-
American Academy of Ophthalmology. Basic and Clinical Science Course (BCSC): Retina and Vitreous. San Francisco, CA: AAO; latest edition.
-
Yanoff M, Duker JS. Ophthalmology. 5th ed. Elsevier; 2019.
-
Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22(2):199–202.
-
Traboulsi EI. Genetic Diseases of the Eye. 2nd ed. Oxford University Press; 2012.
-
Boon CJF, Klevering BJ, Hoyng CB, Keunen JEE, den Hollander AI. Basal laminar drusen and related macular dystrophies. Prog Retin Eye Res. 2010;29(3):213–235.
-
Querques G, Souied EH. Autosomal dominant drusen: clinical spectrum and imaging findings. Surv Ophthalmol. 2014;59(3):238–252.

{kind=link}