
Case Study
A 17-year-old male presented with a long-standing history of difficulty seeing in dim illumination since early childhood.
He reported markedly delayed dark adaptation after moving from brightly lit environments to darkness but denied significant daytime visual problems.
There was no history of photophobia, metamorphopsia, or progressive vision loss. His family history revealed that a sibling had similar symptoms.
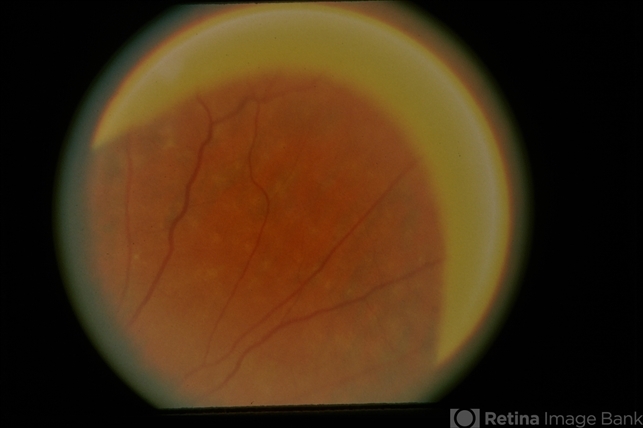
Best-corrected visual acuity was 20/20 in both eyes. Anterior segment examination was unremarkable. Fundus examination demonstrated numerous small, discrete, yellow-white punctate lesions distributed throughout the posterior pole and mid-peripheral retina, sparing the fovea.
Optical coherence tomography (OCT) showed subtle abnormalities at the level of the photoreceptor outer segments and retinal pigment epithelium (RPE).
Standard electroretinography (ERG) performed after conventional dark adaptation showed markedly reduced rod responses.
However, after prolonged dark adaptation of approximately three hours, rod responses improved significantly and approached normal amplitudes.
Based on the clinical presentation, imaging findings, and electrophysiologic testing, a diagnosis of Fundus Albipunctatus was made.
Disease Entity
Fundus albipunctatus is a rare, inherited form of congenital stationary night blindness characterized by numerous small white or yellowish retinal flecks distributed throughout the posterior pole and mid-peripheral retina.
The condition is nonprogressive and primarily affects rod photoreceptor function due to abnormalities in the visual cycle.
The disease is most commonly inherited in an autosomal recessive pattern and is typically associated with mutations in the RDH5 gene, which encodes 11-cis retinol dehydrogenase, an enzyme involved in the regeneration of 11-cis retinal in the visual cycle.
This enzyme is located in the retinal pigment epithelium and plays a critical role in phototransduction.
Fundus albipunctatus is classified among the white dot retinal disorders, but unlike inflammatory white dot syndromes, it represents a hereditary metabolic dysfunction affecting the photoreceptor–RPE interface.
Pathophysiology
The underlying defect in fundus albipunctatus involves impaired regeneration of 11-cis retinal, an essential chromophore required for phototransduction in rod photoreceptors.
Under normal conditions, the visual cycle converts all-trans retinal generated during phototransduction back into 11-cis retinal, which recombines with opsin to form functional rhodopsin.
This process occurs primarily within the retinal pigment epithelium and involves several enzymatic steps.
Mutations in the RDH5 gene disrupt the conversion of 11-cis retinol to 11-cis retinal, resulting in delayed regeneration of rhodopsin.
Consequently, rod photoreceptors require extended periods of dark adaptation before normal function is restored.
This biochemical defect leads to:
-
Delayed rod photoreceptor recovery
-
Accumulation of retinoid byproducts
-
Formation of characteristic subretinal white deposits
Despite impaired rod function, cone photoreceptors typically remain relatively preserved, explaining the generally normal visual acuity and color vision observed in affected individuals.
Epidemiology
Fundus albipunctatus is an uncommon inherited retinal disorder. Precise prevalence estimates are limited due to its rarity and frequent underdiagnosis.
Key epidemiologic features include:
-
Inheritance pattern: Autosomal recessive in most cases
-
Typical age of onset: Early childhood
-
Sex distribution: Equal in males and females
-
Ethnic distribution: Reported worldwide, with several familial clusters identified in East Asian and European populations
Symptoms are usually recognized during childhood or adolescence when patients report difficulty seeing in low-light environments.
Because the condition is stationary rather than progressive, many patients remain stable throughout life.
Clinical Features
The hallmark clinical symptom of fundus albipunctatus is night blindness (nyctalopia) with markedly delayed dark adaptation.
Common symptoms include:
-
Lifelong night blindness
-
Delayed visual recovery after exposure to bright light
-
Difficulty navigating in dim illumination
Importantly, many aspects of visual function remain preserved:
-
Normal or near-normal visual acuity
-
Preserved color vision
-
Minimal or absent visual field loss
Patients often adapt behaviorally by avoiding poorly illuminated environments.
Unlike many inherited retinal dystrophies, progressive visual decline is uncommon, although rare cases associated with macular involvement have been reported.
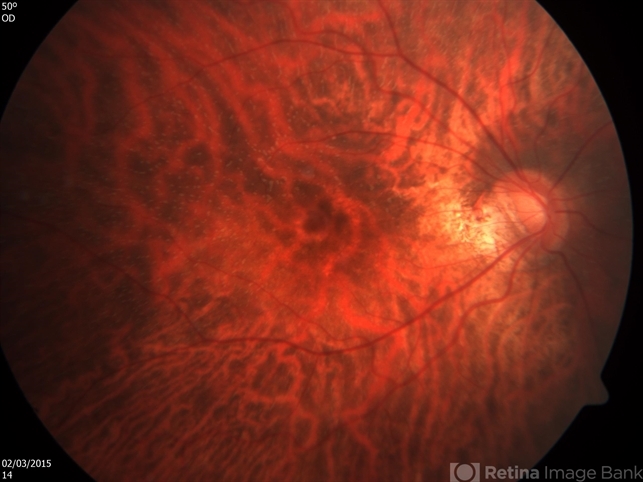
Examination Findings
Fundus examination typically reveals numerous distinctive lesions.
Funduscopic Findings
Characteristic features include:
-
Multiple small yellow-white dots
-
Round or oval lesions
-
Distribution throughout the posterior pole and mid-periphery
-
Relative sparing of the fovea
The lesions are usually located at the level of the retinal pigment epithelium.
Optical Coherence Tomography (OCT)
OCT may demonstrate:
-
Hyperreflective deposits at the level of the RPE
-
Mild disruption of the photoreceptor outer segments
-
Generally preserved foveal architecture
Fundus Autofluorescence
Autofluorescence imaging may reveal:
-
Variable hyperautofluorescent flecks
-
Areas corresponding to retinoid accumulation
Fluorescein Angiography
Fluorescein angiography often shows:
-
Minimal or absent leakage
-
Subtle hyperfluorescent spots corresponding to flecks
Electroretinography (ERG)
ERG findings are diagnostic:
-
Markedly reduced rod responses after standard dark adaptation
-
Substantial recovery after prolonged dark adaptation (2–3 hours)
This delayed recovery is a key distinguishing feature.
Differential Diagnosis
Several inherited retinal disorders and white dot conditions may resemble fundus albipunctatus.
Important differential diagnoses include:
Retinitis punctata albescens
-
Progressive rod-cone dystrophy
-
Persistent ERG abnormalities even after prolonged dark adaptation
-
Gradual visual field loss
Stargardt disease
-
Macular atrophy
-
Pisciform flecks
-
Decreased central vision
White dot syndromes
Examples include:
-
Multiple Evanescent White Dot Syndrome (MEWDS)
-
Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
These are inflammatory and typically present with acute visual symptoms.
Vitamin A deficiency
-
Night blindness
-
Reversible with vitamin A supplementation
Careful electrophysiologic testing helps distinguish these conditions.
Diagnosis
The diagnosis of fundus albipunctatus is based on a combination of clinical, imaging, and electrophysiologic findings.
Key diagnostic elements include:
-
Lifelong night blindness
-
Characteristic punctate retinal flecks
-
Normal visual acuity
-
Delayed rod recovery on ERG
Genetic testing can confirm mutations in:
-
RDH5 (most common)
-
Rarely other visual cycle genes
Multimodal imaging—including OCT and fundus autofluorescence—supports the diagnosis and helps exclude other retinal dystrophies.
Management
There is currently no definitive cure for fundus albipunctatus.
Management is largely supportive and focused on monitoring.
Observation
Most patients require only periodic ophthalmic evaluation to monitor for rare complications.
Genetic Counseling
Because the disease is inherited, genetic counseling is recommended for affected families.
Vitamin A Derivatives
Experimental studies have investigated 9-cis retinal supplementation and related compounds to bypass the biochemical defect in the visual cycle.
Some patients show improvement in dark adaptation, though these treatments remain investigational.
Low-Vision Support
In rare cases with functional limitations, adaptive strategies such as improved lighting conditions may be helpful.
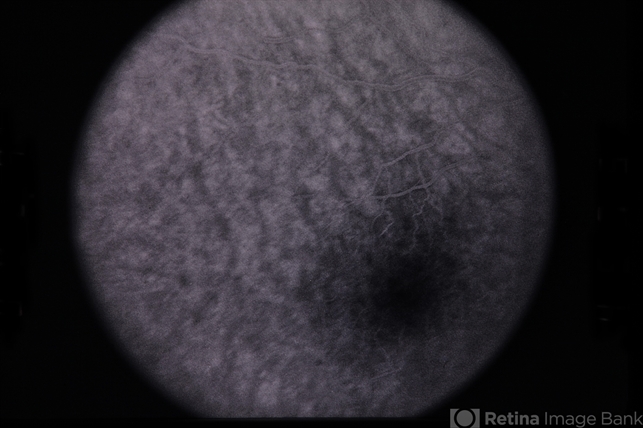
Prognosis
The prognosis for fundus albipunctatus is generally excellent.
Important prognostic features include:
-
Stable visual acuity throughout life
-
Nonprogressive disease course
-
Preserved cone function
Night blindness typically persists but does not worsen significantly over time.
Rarely, some patients may develop macular involvement or cone dysfunction later in life, though this is uncommon.
With appropriate counseling and monitoring, most patients maintain good visual function.
References
-
Ryan SJ, Schachat AP, Wilkinson CP, et al. Retina. 6th ed. Elsevier; 2018.
-
Yanoff M, Duker JS. Ophthalmology. 5th ed. Elsevier; 2019.
-
American Academy of Ophthalmology. Basic and Clinical Science Course (BCSC): Inherited Retinal Diseases and Degenerations. AAO; 2023.
-
Weleber RG. Fundus albipunctatus and related disorders of the visual cycle. In: Ryan SJ, ed. Retina. Elsevier; 2018.
-
Thompson DA, Gal A. Genetic defects in the visual cycle and congenital stationary night blindness. Prog Retin Eye Res. 2003;22(5):683–703.
-
Sergouniotis PI, et al. RDH5 mutations and the clinical spectrum of fundus albipunctatus. Invest Ophthalmol Vis Sci. 2011;52(7):4758–4766.

{kind=link}