
Case Study
A 32-year-old female presents with a one-week history of sudden-onset shimmering lights (photopsias) and blurred vision in her left eye.
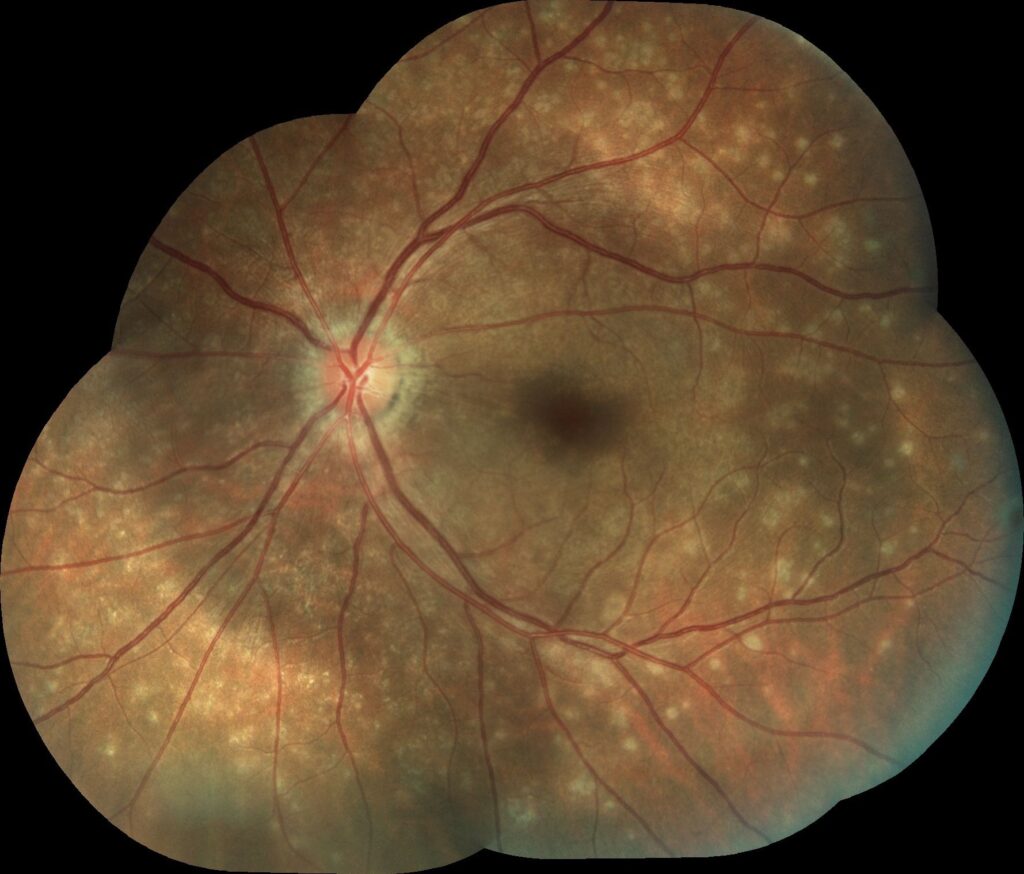
She reports experiencing flu-like symptoms approximately two weeks prior. She denies any history of ocular trauma, surgery, or significant systemic illness, although she notes being moderately myopic.
On examination, her best-corrected visual acuity is 6/12 in the left eye and 6/6 in the right eye. Slit-lamp examination of the anterior segment is unremarkable, with no signs of anterior chamber inflammation. Intraocular pressures are normal bilaterally.
Dilated fundus examination of the left eye reveals multiple, subtle, small, greyish-white dots scattered at the level of the outer retina and retinal pigment epithelium (RPE), predominantly in the posterior pole and mid-periphery.
There is also mild optic disc edema and foveal granularity. The right eye fundus appears normal.
Optical Coherence Tomography (OCT) shows disruption of the ellipsoid zone (EZ) and interdigitation zone (IZ) corresponding to the white dots and foveal granularity.
Fundus autofluorescence (FAF) demonstrates faint hyperautofluorescent spots corresponding to the lesions. Fluorescein angiography (FA) reveals early punctate hyperfluorescence with late staining of the lesions and optic disc leakage.
Indocyanine green angiography (ICGA) shows multiple hypofluorescent spots in the mid and late phases.
A diagnosis of Multiple Evanescent White Dot Syndrome (MEWDS) is made based on the characteristic clinical presentation and multimodal imaging findings.
Disease Entity
White Dot Syndromes (WDS) encompass a heterogeneous group of idiopathic inflammatory conditions primarily affecting the outer retina, RPE, choroid, and occasionally the inner retina.
These syndromes are characterized by the presence of multifocal, discrete, whitish or yellowish lesions at the posterior pole and/or periphery.
While often grouped together due to overlapping clinical features, each syndrome possesses distinct characteristics regarding demographics, clinical course, imaging findings, and prognosis.
Commonly recognized entities within the WDS spectrum include:
- Multiple Evanescent White Dot Syndrome (MEWDS)
- Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
- Birdshot Chorioretinopathy (BCR)
- Multifocal Choroiditis and Panuveitis (MCP)
- Punctate Inner Choroidopathy (PIC)
- Serpiginous Choroidopathy (SC)
- Acute Zonal Occult Outer Retinopathy (AZOOR)
- Acute Retinal Pigment Epitheliitis (ARPE)
These conditions typically affect young, otherwise healthy adults and often present with symptoms like photopsias, floaters, blurred vision, scotomas (blind spots), or decreased night vision.
Differentiating between these syndromes is crucial for appropriate management and prognostication.
Pathophysiology
The exact etiology and pathophysiology of most White Dot Syndromes remain elusive, though immune-mediated mechanisms and infectious triggers (particularly viral) are strongly implicated.
Many cases are preceded by a viral prodrome, suggesting an initial infectious event followed by an aberrant autoimmune response targeting retinal or choroidal antigens.
- Immune Dysregulation: An underlying autoimmune process is suspected in many WDS. Genetic predisposition, such as the strong association of Birdshot Chorioretinopathy with HLA-A29, supports this hypothesis. In conditions like MCP and PIC, choroidal neovascularization (CNV) can develop, indicating a chronic inflammatory drive and RPE dysfunction.
- Infectious Triggers: Viral infections (e.g., Epstein-Barr virus, Coxsackievirus, influenza) are often reported preceding the onset of MEWDS and APMPPE. It is hypothesized that these infections might trigger a cross-reactive immune response against retinal/choroidal components or cause direct viral-mediated damage.
- Choroidal Ischemia: In APMPPE, the characteristic placoid lesions are thought to result from inflammation and occlusion of the choriocapillaris, leading to ischemic damage of the overlying RPE and outer retina. ICGA findings often support primary choroidal involvement.
- RPE/Photoreceptor Involvement: MEWDS is characterized by transient dysfunction primarily at the level of the photoreceptors and RPE, as evidenced by the evanescent nature of the lesions and OCT findings showing disruption of the outer retinal layers. The exact target antigen remains unknown.
Research continues to explore the complex interplay between genetic susceptibility, environmental triggers (like infections), and immune pathways in the development of these diverse syndromes.
Epidemiology
The epidemiology varies significantly among the different White Dot Syndromes:
- MEWDS: Typically affects young adult females (average age 20-40 years), often with myopia. It is usually unilateral and has an excellent prognosis with spontaneous resolution.
- APMPPE: Affects young adults (20s-30s) with no gender predilection. Often bilateral, though potentially asymmetric. Frequently associated with a preceding viral illness. Prognosis is generally good, but visual recovery can be prolonged, and recurrences are possible.
- Birdshot Chorioretinopathy (BCR): Primarily affects middle-aged Caucasians (40s-60s) and shows a very strong association with the HLA-A29 antigen (>95%). It is typically bilateral and chronic, often leading to significant visual impairment if untreated due to complications like cystoid macular edema (CME) and CNV.
- Multifocal Choroiditis and Panuveitis (MCP): Predominantly affects young myopic females (20s-40s). Usually bilateral and characterized by chronic or recurrent inflammation involving the choroid and retina, often with anterior chamber and vitreous inflammation (panuveitis). High risk of CNV and subretinal fibrosis.
- Punctate Inner Choroidopathy (PIC): Similar demographics to MCP (young myopic females). Bilateral involvement is common. Characterized by smaller, punctate lesions primarily at the posterior pole, with minimal vitritis. Also carries a high risk of CNV.
- Serpiginous Choroidopathy (SC): Affects adults typically between 30-60 years, with no clear gender predilection. Usually bilateral but often asymmetric. Characterized by geographic, serpentine-like lesions spreading centrifugally from the optic disc or macula. Can be associated with tuberculosis in some regions.
- AZOOR: Primarily affects young to middle-aged females. Characterized by acute loss of visual field in one or more zones, often associated with photopsias, and minimal visible fundus changes initially (occult). Can be progressive.
The overall incidence of WDS is low, but they represent an important group of inflammatory eye diseases due to their potential for causing visual morbidity, particularly the chronic forms like BCR, MCP, PIC, and SC.
Clinical Features
Symptoms vary depending on the specific syndrome, but often include:
- Photopsias: Flashing or shimmering lights, common in MEWDS and AZOOR.
- Floaters: New or increased floaters, often indicating vitritis (common in MCP, BCR, SC).
- Blurred Vision: Ranging from mild to severe, depending on macular involvement, CME, or CNV.
- Scotomas: Blind spots in the visual field, corresponding to areas of retinal/choroidal damage. Paracentral scotomas are typical in PIC and MCP; zonal field loss in AZOOR; enlarged blind spot in MEWDS.
- Nyctalopia: Decreased night vision, a prominent feature in BCR.
- Metamorphopsia: Distortion of vision, often associated with CME or CNV.
- Pain/Photophobia: Less common than in anterior uveitis, but can occur, especially with significant inflammation.
Onset is typically acute or subacute. Unilateral presentation is characteristic of MEWDS, while most others (APMPPE, BCR, MCP, PIC, SC, AZOOR) tend to be bilateral, although often asymmetric at onset.
Examination Findings
Slit-lamp examination may reveal anterior chamber cells and flare or keratic precipitates in syndromes with anterior segment involvement (MCP, BCR, SC).
Vitreous cells are common in MCP, BCR, and SC, but are typically absent or minimal in MEWDS, APMPPE, PIC, and AZOOR.
Funduscopic Findings:
- MEWDS: Multiple, small (100-200 µm), discrete, greyish-white dots at the RPE/outer retinal level, mainly posterior pole; often evanescent (disappear within weeks). Foveal granularity and mild optic disc edema are common.
- APMPPE: Creamy-yellow, placoid (plate-like) lesions at the RPE level, varying in size, scattered throughout the posterior pole. Lesions evolve, fading over weeks to leave areas of RPE atrophy.
- BCR: Multiple, ovoid, cream-colored choroidal lesions (“birdshot spots”), typically 1/4 to 1/2 disc diameter, radiating from the optic disc, most prominent nasally. Vitritis is almost always present. Optic disc edema and CME are common.
- MCP/PIC: Multiple, discrete, punched-out, yellow-white chorioretinal lesions of varying size (50-1000 µm), primarily at the posterior pole and mid-periphery. Active lesions may appear creamy. Vitritis is common in MCP, minimal in PIC. Peripapillary atrophy and CNV are frequent complications.
- SC: Geographic, yellowish-white lesions with irregular (serpiginous) borders, typically starting near the optic disc and spreading centrifugally. Active edges appear creamy, while healed areas show RPE atrophy and choriocapillaris loss.
- AZOOR: Fundus may appear normal initially or show subtle RPE changes. Later, zonal RPE atrophy and pigmentary changes develop, corresponding to the visual field defects.
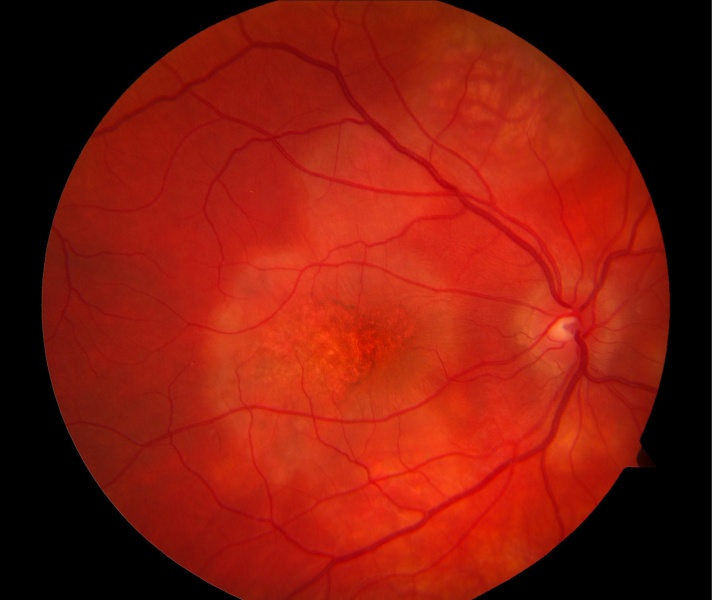
Differential Diagnosis
The differential diagnosis for white lesions in the fundus is broad and includes infectious, inflammatory, neoplastic, and degenerative conditions:
- Infectious Chorioretinitis: Syphilis, tuberculosis, toxoplasmosis, cytomegalovirus (CMV), herpes viruses, fungal infections (Candida, Aspergillus), diffuse unilateral subacute neuroretinitis (DUSN).
- Other Inflammatory Conditions: Sarcoidosis, Vogt-Koyanagi-Harada (VKH) disease, sympathetic ophthalmia, primary intraocular lymphoma (masquerade syndrome).
- Degenerative/Other: Pattern dystrophies, fundus albipunctatus, Bietti crystalline dystrophy, talc retinopathy.
Distinguishing WDS from infectious causes is paramount, as immunosuppressive therapy appropriate for WDS can worsen infections.
Diagnosis
Diagnosis is primarily clinical, based on characteristic history, symptoms, and funduscopic findings, supported by multimodal imaging.
Key Investigations:
- Fundus Photography: Documents lesion appearance, distribution, and evolution.
- Fundus Autofluorescence (FAF): Highly valuable for assessing RPE health. Active lesions often show variable FAF (hyper- or hypo-), while atrophic areas are typically hypoautofluorescent. MEWDS lesions are often hyperautofluorescent.
- Optical Coherence Tomography (OCT): Essential for detecting subtle outer retinal/RPE disruption (e.g., EZ/IZ loss in MEWDS, APMPPE), intraretinal/subretinal fluid, CME, and CNV.
- Fluorescein Angiography (FA): Helps characterize lesions. APMPPE shows early blockage and late staining. MEWDS shows early punctate hyperfluorescence with late staining (“wreath-like” pattern) and disc leakage. MCP/PIC lesions show early blockage or hyperfluorescence with late staining. Useful for detecting CNV and CME.
- Indocyanine Green Angiography (ICGA): Visualizes choroidal circulation. Often shows hypofluorescent spots corresponding to lesions in MEWDS, APMPPE, MCP, PIC, and BCR, suggesting primary choroidal inflammation or ischemia.
- Visual Field Testing: Crucial for detecting and monitoring scotomas (e.g., enlarged blind spot in MEWDS, central/paracentral defects in PIC/MCP, zonal loss in AZOOR, generalized constriction in BCR).
- Electroretinography (ERG): Can be abnormal, particularly in AZOOR (reduced photopic/scotopic responses) and BCR (reduced b-wave amplitude).
Laboratory Testing: Generally guided by clinical suspicion to rule out infectious and systemic inflammatory mimics:
- Complete blood count (CBC), Erythrocyte Sedimentation Rate (ESR), C-reactive protein (CRP).
- Syphilis serology (VDRL/RPR, FTA-ABS/TPPA).
- Tuberculosis testing (PPD skin test or Interferon-Gamma Release Assay – IGRA), Chest X-ray.
- Angiotensin-Converting Enzyme (ACE) level, Lysozyme (for sarcoidosis).
- HLA typing (HLA-A29 for suspected BCR).
- Consider testing for other infections (e.g., Bartonella, Lyme) based on exposure history and clinical picture.
Management
Management strategies for White Dot Syndromes vary significantly based on the specific diagnosis, severity, laterality, and presence of complications like CNV or CME.
- Observation: Many cases of MEWDS, APMPPE, and ARPE are self-limiting and resolve spontaneously without treatment, often with good visual recovery. Close monitoring is essential initially.
- Corticosteroids:
- Systemic corticosteroids (oral prednisone) may be considered for severe bilateral APMPPE affecting the fovea, severe MEWDS with significant vision loss, or initial control of inflammation in MCP, PIC, BCR, and SC.
- Periocular or intravitreal corticosteroids can be used for unilateral or asymmetric disease, particularly for managing CME in BCR or MCP.
- Topical corticosteroids are generally not effective for posterior segment inflammation but may be used if there is associated anterior uveitis.
- Immunosuppressive Therapy (IMT): Long-term immunosuppression is often required for chronic, recurrent, or vision-threatening WDS like BCR, MCP, PIC, and SC to control inflammation, prevent damage, and spare long-term steroid use. Common agents include:
- Antimetabolites: Methotrexate, mycophenolate mofetil, azathioprine.
- Calcineurin inhibitors: Cyclosporine, tacrolimus.
- Biologic agents: TNF-α inhibitors (adalimumab, infliximab) are increasingly used, particularly for refractory BCR and MCP.
- Anti-VEGF Therapy: Intravitreal injections of anti-vascular endothelial growth factor agents (e.g., bevacizumab, ranibizumab, aflibercept) are the standard of care for treating choroidal neovascularization (CNV) associated with MCP, PIC, SC, and occasionally BCR.
- Laser Photocoagulation: Rarely used now for CNV due to the success of anti-VEGF therapy, but may have a role in specific cases of extrafoveal CNV.
Management decisions should be individualized, considering the potential benefits and risks of treatment, especially systemic immunosuppression.

Prognosis
The visual prognosis is highly variable among the White Dot Syndromes:
- MEWDS: Generally excellent. Most patients experience complete spontaneous recovery of vision within weeks to months, although mild residual photopsias or visual field defects can persist in some.
- APMPPE: Usually good. Visual acuity typically recovers to near-normal levels, but recovery can take weeks to months. Some patients may have persistent RPE changes or, rarely, develop CNV.
- BCR: Guarded without treatment. It is a chronic, progressive disease. While vision may remain good initially, long-term inflammation often leads to complications like CME, CNV, cataract, glaucoma, and optic atrophy, causing significant visual impairment if not managed with long-term immunosuppression.
- MCP & PIC: Variable to guarded. These conditions have a high rate of CNV development, which is a major cause of vision loss. Subretinal fibrosis can also occur. Prognosis depends on the frequency and severity of recurrences and the response to treatment, including anti-VEGF for CNV.
- SC: Guarded, particularly if the fovea is involved. Lesions progressively enlarge, leading to irreversible RPE and choriocapillaris loss and corresponding scotomas. Foveal involvement often results in significant central vision loss. CNV can also occur.
- AZOOR: Variable. Some patients stabilize, while others experience progressive visual field loss. Central vision may remain good if the fovea is spared.
Conclusion
White Dot Syndromes represent a diverse group of inflammatory chorioretinopathies characterized by multifocal white lesions.
While sharing some overlapping features, distinct clinical patterns, demographics, and multimodal imaging findings allow for specific diagnoses, ranging from self-limiting conditions like MEWDS to chronic, vision-threatening diseases like Birdshot Chorioretinopathy and Serpiginous Choroidopathy.
Accurate diagnosis, aided significantly by FAF, OCT, FA, and ICGA, is crucial for differentiating these syndromes from infectious mimics and guiding appropriate management.
Treatment varies from observation to long-term immunosuppression and anti-VEGF therapy for complications. Prognosis is highly dependent on the specific syndrome and the presence of complications.
Continued research is needed to fully elucidate the pathophysiology and optimize treatment strategies for these challenging conditions.
Would you have interest in taking retinal images with your smartphone?
Fundus photography is superior to fundus analysis as it enables intraocular pathologies to be photo-captured and encrypted information to be shared with colleagues and patients.
Recent technologies allow smartphone-based attachments and integrated lens adaptors to transform the smartphone into a portable fundus camera and Retinal imaging by smartphone.
References
- Mount GR, Kaufman EJ. White Dot Syndromes.
- Reddy AK, Grewal DS, Jaffe GJ. Evaluation and management of white dot syndromes. Surv Ophthalmol. 2022 Nov-Dec;67(6 ):1719-1742. doi: 10.1016/j.survophthal.2022.05.003. Epub 2022 May 19. PMID: 35598671.
- Raven ML, Ringeisen AL, Yonekawa Y, Stem MS, Faia LJ, Gottlieb JL. Multi-modal imaging and anatomic classification of the white dot syndromes. Int J Retina Vitreous. 2017 Jul 11;3:12. doi: 10.1186/s40942-017-0069-y. PMID: 28702257; PMCID: PMC5504820.
- Mrejen S, Sarraf D, Chexal S, et al. Multimodal imaging of multifocal choroiditis. Retina. 2014 May;34(5):856-65. doi: 10.1097/IAE.0000000000000018. PMID: 24743642.

{kind=link}