
CASE REPORT
A 32-year-old male software engineer presented to an ophthalmology clinic with complaints of progressively worsening vision, including increased difficulty in reading small print and occasional blurring of vision, particularly in low-light conditions.

On examination, visual acuity was 20/50 in the right eye and 20/60 in the left eye, with normal intraocular pressure.
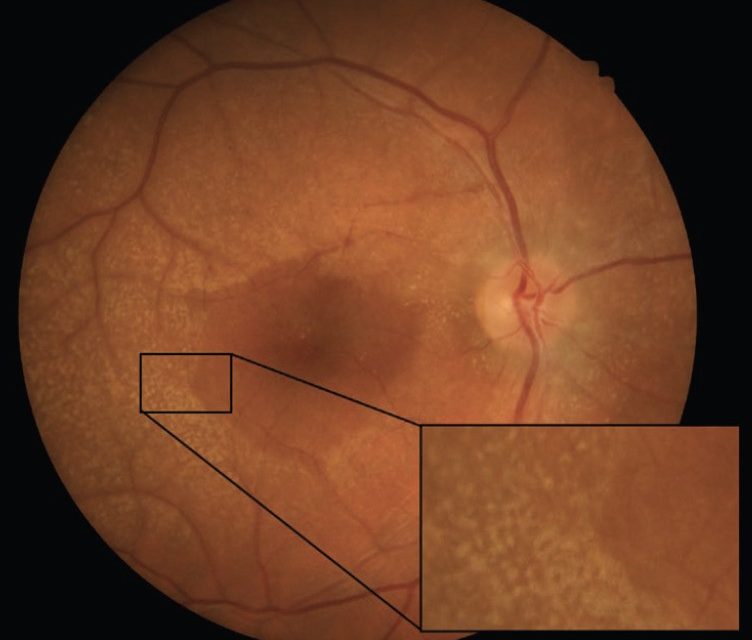
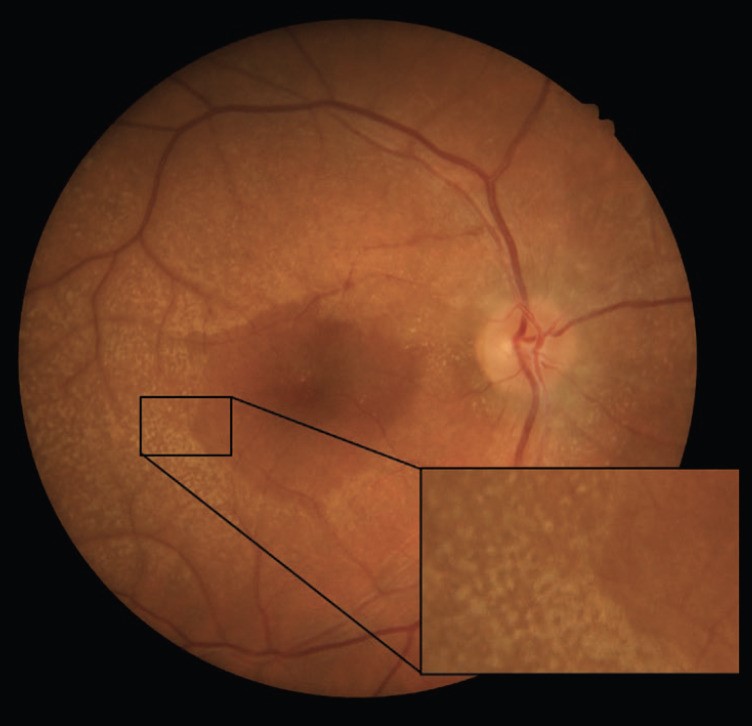
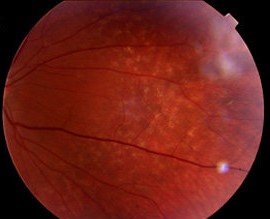
Anterior segment examination revealed no remarkable findings, while posterior segment examination showed bilateral dot and fleck retinopathy with areas of retinal thinning and irregular pigmentation.
Optical Coherence Tomography (OCT) confirmed irregularities in the retinal pigment epithelium (RPE) and disruption of the ellipsoid zone in the macular region, consistent with the diagnosis of Alport Syndrome.
DISEASE
Alport syndrome is a genetic disorder. It is identified by kidney disease, hearing loss, and eye abnormalities.
Alport syndrome is a hereditary glomerular basement membrane disease as a result of mutations in the genes COL4A3/4/5.
These genes encode the type IV collagen alpha 3-5 chains. The most frequent inheritance is X-linked dominant, followed by autosomal recessive and autosomal dominant.
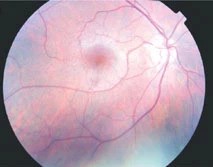
The characteristic ocular features of Alport syndrome are corneal opacities, anterior lenticonus, and cataracts, central perimacular and peripheral coalescing fleck retinopathies, and temporal retinal thinning.
Rarely, posterior polymorphous corneal dystrophy, a macular hole, or a maculopathy impairs vision. Lenticonus, corneal dystrophy, central and peripheral fleck retinopathies, temporal retinal thinning, and giant macular holes are all highly suspicious for the diagnosis of Alport syndrome.
Physical examination
Slit-lamp:
- The most frequent ocular signs:
- Anterior lenticonus with “oil droplet” sign and usually axial, 2–7 mm.
- Dot-and-flecks retinopathy with “lozenge” sign (dull macular reflex): it’s seen like white or yellow granulations that are superficially located.
- Less common:
- corneal posterior polymorphous dystrophy: endotelial vesicles are seen in the form of clusters (“dougnuts) or bands (“snail tracks”)
- secondary calcifications of hypercalcemia (if renal failure) in conjunctiva and sclera.
- corneal erosions are seen in the anterior cornea.
- corneal opacities.
- microcornea.
- cataract.
- posterior lenticonus.
- spontaneous capsule rupture.
- lammellar macular holes.
- disturbances of foveal pigmentation, including a bull’s eye or vitelliform maculopathy.
- temporal macular thinning.
Ocular symptoms:
- Acute episodes of pain, watering, photophobia, and blurred vision caused by corneal erosions.
- Progressive visual loss caused by corneal posterior polymorphous dystrophy.
- Lenticular myopia caused by anterior lenticonus.
- Astigmatism.
- Decreased acuity visual caused by cataract.
- Central visual loss caused by macular holes. They respond poorly to surgery.
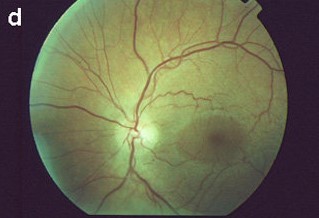
Retinopathy:
The α3α4α5 type IV collagen network is present in the normal ILM and in the retinal pigment epithelium basement membrane of Bruch’s membrane.
In Alport syndrome, the ILM/nerve fiber layer and Bruch’s membrane are both thinned. The thinning of these membranes contributes to the appearance of retinopathy and macular holes.
The dot-and-fleck retinopathy is seen like white or yellow granulations that are superficially located with a “lozenge” sign (dull macular reflex as a result of the demarcation between the perimacular retinopathy and the thinned macula).
Besides, it presents a hyperreflectivity on OCT of the ILM/nerve fiber layer in the distribution of the nerve fiber layer corresponding with the location of the retinopathy.
A thinned ILM may be more susceptible to tractional forces from the vitreous, interfere with the transport of nutrients, or impair the clearance of waste products, being all of them the origin of dot-and-flecks retinopathy.
The central retinopathy varies from scattered whitish-yellow dots and flecks to a dense, almost confluent annulus around the region of temporal retinal thinning.
The central and peripheral retinopathies seem to have an identical pathogenesis in X-linked and autosomal recessive Alport syndrome.
Retinopathy has a prognostic value in determining early-onset renal failure if present. Fundus albipunctatus, other causes of generalized drusen and pigmentary retinopathies, that are usually accompanied by night blindness and progressive visual loss, should be considered in retinopathy differential diagnosis.
Dot-and-fleck retinopathy and temporal macular thinning do not affect vision. On the other hand, macular holes affect central vision and have a poor response to surgery.
Corneal Pathology:
- Recurrent erosions: due to defective adhesion between epithelium and Bowman’s membrane. They typically occur in individuals with early-onset renal failure and extrarenal features. Sometimes, corneal erosions are found in different members of the same family, but they do not appear to be associated with specific mutations.
- Posterior polymorphous corneal dystrophy: the cause is similar to recurrent erosions, and defective adhesion between layers. It can be asymptomatic or patients complaining about gritty, watering eyes and photophobia. Some advanced cases may require corneal transplantation. The other ones are just symptomatic treatments. It can be detected by slit-lamp examination, confocal microscopy, or anterior segment Optical Coherence Tomography.
Management
REFERENCES
- Simon Watson, Bush Jeffrey S. Alport Syndrome. StatPearls Publishing. 2018 jan.
- Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F: Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol 24: 364–375, 2013.
- Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D.Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015; 10(4):703-9.
- Fawzi AA, Lee NG, Eliott D, Song J, Stewart JM. Retinal findings in patients with Alport syndrome: expanding the clinical spectrum. Br J Ophthalmol. 2009; 93:1606–1611.
- Zhang Y, Ding J.Renal, auricular and ocular outcomes of Alport syndrome and their current management. Pediatr Nephrol. 2017.
- Savige J, Liu J, DeBuc DC, Handa JT, Hageman GS, Wang YY, Parkin JD, Vote B, Fassett R, Sarks S, Colville D: Retinal basement membrane abnormalities and the retinopathy of Alport syndrome. Invest Ophthalmol Vis Sci. 2010; 51: 1621–1627.
RETINAL IMAGING BY YOUR SMARTPHONE

RETINAL IMAGING BY YOUR SMARTPHONE
{kind=link}